
|
|
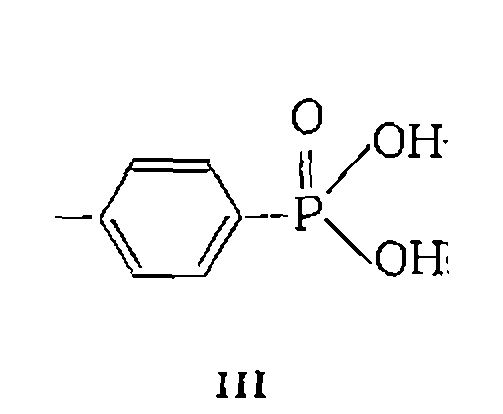
ИММОБИЛИЗОВАННЫЕ ГОМОГЕННЫЕ КАТАЛИЗАТОРЫНесмотря на высокую активность и селективность, применение гомогенных катализаторов в промышленности ограничивается трудностями, связанными с отделением их от реакционной смеси и последующей регенерацией. Объединение преимуществ гомогенного и гетерогенного катализа может быть достигнуто «ге- терогенизацией» или «иммобилизацией» гомогенных катализаторов путем их связывания с твердым носителем. Закрепление гомогенных катализаторов на нерастворимой в реакционной массе основе осуществляется химическим связыванием с носителем, заполнением пор носителя раствором гомогенного катализатора или включением гомогенного катализатора в гель. Наибольшее распространение получило химическое связывание гомогенных катализаторов с органическими или неорганическими носителями. В качестве органического носителя обычно используют полистирол и его сополимеры с сшивающими агентами (дивинилбензол, бутадиен), которые обеспечивают образование трехмерной пространственной сетки полимера, нерастворимого в воде и органических растворителях. Применяют также различные полиамиды, поливинилпиридин и полиакриловую кислоту, сшитые дивинилбензолом, и т. п. Органический полимер с закрепленными каталитически активными группами может использоваться в виде геля, не имеющего внутренней поверхности, или иметь развитую внутреннюю поверхность (20— 70 м2/г) и сеть крупных пор разного диаметра (10—70 нм). Размер пор и общая пористость регулируются в процессе синтеза полимера изменением количества сшивающего агента и добавками инертного растворителя. Химическое связывание кислотных или основных групп с органическим полимерным .носителем либо полимеризация мономеров, содержащих такие группы, приводит к образованию ионообменных смол. Их .используют, для выделения различных катионов или анионов из раствора’за счет ионного обмена, а также в качестве кислотных, основных или нуклеофильных; катализаторов. Для кислотного катализа представляют интерес катионообменные смолы (катиониты), содержащие сильнокислотные группы ■ листирольные (I — катиониты КУ-2, СДВ-3, СБС и др.) и фосфорсодержащие (II—катиониты РФ и III — катиониты СФ„ КФ), содержащие группы
Содержание кислотных групп в катионите (так называемая обменная емкость) составляет для сульфополистирольных катионитов 3—5 мэкв/г, для фосфорсодержащих 7—10 мэкв/г. Перечисленные катиониты эффективно катализируют многие реакции, подверженные гомогенному кислотному катализу. К ним; относятся гидратация третичных непредельных углеводородов (изобутена, изопентеиа), этерификация карбоновых кислот, ал- килирование ароматических соединений олефинами, реакция формальдегида с олефинами (реакция Принса) и т. п. Высокое содержание кислотных групп в реакционной массе, которого, можно достичь применением катионитов, в ряде случаев обеспечивает значительно более высокую скорость реакции по сравнению с гомогенным катализом. Вместе с тем применение катионитов ограничивается температурой: 140 °С для КУ, СДВ, СБС, 150 °С для СФ, КФ, 80 °С для РФ, выше которой начинается разложение (термическая деструкция) с отщеплением каталитически активных групп. Для основного и нуклеофильного катализа используют анио- иО'Обмениые смолы (аниониты), содержащие группы
пиридине. Аниониты, в особенности ионогенные с группами
основно-каталитнческие реакции альдольной конденсации и др. Роль нуклеофильных катализаторов могут выполнять и другие анионы, связанные с анионитом путем обмена с гидроксил-ионом, например:
Органические полимерные носители широко используют ь последнее время и для "иммобилизации металлокомплексных катализаторов. Одним из путей для этого является ионный обмен с ионитами. Такой способ пригоден для связывания заряженных комплексов металлов. В частности, гетерогенизация комплекса Катион сульфокатионитом:
Полученный гетерогенный катализатор проявляет те же каталитические свойства, что и гомогенный, в реакции карбони- лирования аллилхлорида: Иммобилизадия нейтральных комплексов металлов осуществляется путем связывания их с закрепленными на носителе лигандами. Для этой цели используют модифицированный фосфи- новыми группами полистирол
с группами са металла происходит при замещении низкомолекулярного растворимого лиганда высокомолекулярным, например: Иммобилизованный таким путем комплекс родия, аналогично исходному гомогенному, проявляет высокую каталитическую активность в реакциях гидрирования олефинов. Таким же способом получают гетерогенизированные катализаторы гидрофор- милирования карбонилирования метанола Наряду с органическими полимерными носителями для иммобилизации комплексов металлов используют минеральные' (алюмосиликаты, силикагель). Их главное преимущество по сравнению с органическими состоит в повышенной термической стойкости и механической прочности. Для гетерогенизации комплекса металла к ОН-группам силикагеля прививают крем- нийорганическую «ножку» с лигандом, например, по реакции
Последующий лигандный обмен с комплексами металлов позволяет получить всевозможные иммобилизованные катализаторы гидрирования, гидроформилирования, карбонилирования. Другой способ гетерогенизации на минеральных носителях состоит в непосредственном взаимодействии комплекса металла с ОН- или SiO-группами носителя:
Полученный комплекс циркония является активным катализатором полимеризации олефинов. Комплексы с аналогичными свойствами получают также путем взаимодействия
Гетерогенизация металлокомплексных катализаторов на неорганических носителях расширяет возможности такого типа катализаторов, позволяя использовать их в катализе высокотемпературных газофазных процессов, неосуществимых из-за термодинамических ограничений при относительно- низких температурах в растворах. Это касается, в частности, важных про: мышленных процессов дегидрирования, дегидроконденсации и дегидроциклизации. Примером эффективного катализатора дегидрирования углеводородов являемся комплекс гетерогенизированный на оксиде алюминия. Иммобилизация гомогенных катализаторов путем заполнения пор носителя раствором катализатора проводится по методике, аналогичной способу приготовления неподвижных сЬяз лля газожидкостной хроматографии. Например, раствор парения метанола получают носитель, поры которого заполнены раствором катализатора в гликоле. Такой катализатор, как и гомогенный, активен в реакциях гидрирования и изомеризации олефинов. Аналогично получают катализаторы гидроформилиро- вания.например
Метод гетерогенизации гомогенных катализаторов включением их в каркас полимерного геля используют в основном в химической энзимологии для закрепления (иммобилизации) ферментов. Этот же метод был успешно использован для гетерогенизации металлокомплексных катализаторов полимеризации олефинов. Для этой цели использовали сополимеры бутадиена с винилпиридином, метакриловой кислотой, натуральный или синтетический каучуки, в которые вводили компоненты гомогенной каталитической системы Циглера — Натта. Включенные в гель комплексы проявили высокую каталитическую активность и стабильность, 'обеспечивающую суммарный выход полиэтилена до 0,5 кг на 1 г титана, что значительно превышает ■соответствующие показатели гомогенного катализатора. Кроме указанных преимуществ иммобилизованных гомогенных хатализаторов важной их особенностью является возможность гетерогенизации на единой основе разных активных групп, катализирующих последовательные стадии одного процесса. При этом допустимы комбинации, невозможные в гомогенных условиях, например одновременный катализ кислотой и основанием при использовании смеси катионита и анионита. Эта смесь катализирует, в частности, деполимеризацию паральдеги- да (кислотный катализ) и последующую альдольную конденсацию уксусного 1альдегида (основный катализ). Полифункцио- нальный катализатор, содержащий привитые к полистиоольной матрице группы позволяет осуществить в одну стадию промышленно важный «альдокс» процесс, который включает следующие реакции: гид- роформилирование пропилена (катализатор Rh), альдольная конденсация образующихсямасляного н изома-сляного альдегидов (основный катализ Кинетические закономерности реакций, катализируемых включенными в гель катализаторами, растворами катализатора в порах носителя и гелевыми ионитами подобны закономерностям гетерофазных реакций (гл. V), а катализ пористыми ионитами и катализаторами, закрепленными на поверхности неорганических носителей, аналогичен гетерогенному катализу (гл. VI). В обоих случаях процесс характеризуется наличием стадий диффузии реагентов из потока к частице катализатора, диффузии реагентов внутри частицы катализатора к каталитически активным группам, химической реакций и обратной диффузии продуктов реакции в поток реагентов. (Математическое описание подобных процессов приведено в гл. V и VI; там же рассмотрены закономерности изменения их селективности по сравнению с гомогенными процессами.) Вопросы и упражнения 1. Для реакции
Каков будет вид уравнения нри избытке этилеиоксида по отношению к катализатору и при разных соотношениях величин 2. Реакция протекает но схеме нуклеофильного катализа (III-2). При условии 3. Выведите кинетическое уравнение и предложите метод обработки кинетических данных по реакции этилеиоксида с меркаптаном (с. 151), если все кинетические эксперименты нроведены при соизмеримых концентрациях исходных реагентов. 4. Реакция
Выведите кинетическое уравнение, если и 5. Реакция а-метилниридина (А) с формальдегидом при катализе щелочью дает 6. Выведите кинетическое уравнение для реакции копденсации нитросо- сдинения с кетоном при катализе щелочью:
если известно, что скорость лимитируется стадией атаки молекулы кетона карбаниоиом нитросоединения, образующимся под действием щелочи по обратимой реакции, константа равновесия которой достаточно велика. 7. Если реакция А-—катализируется разбавленными водными растворами как кислот, так и оснований, две прямые зависимости константы скорости от кислотности среды в логарифмических координатах нересекаются в одной точке (изокаталитической). Покажите, как зависит ее положение от констант скорости кислотно-оснбвно-каталитических реакций. 8. Найдите коистапту основности 9. Выведите кинетическое уравнение кислотно-каталитнческой реакции оксимирования ацетона, протекающей по схеме
если первая стадия катализируется кислотой и параллельно протекает с достаточно высокой скоростью без катализатора, а степень лротонирования промежуточного продукта на второй стадии мала. Как связано положение максимума 10. Реакция гидролиза кеталя R'R"C(OR)2 в водном растворе серной кислоты характеризуется следующей зависимостью скорости от кислотности:
Каков механизм реакции? Участвует ли молекула воды в лимитирующей стадии гидролиза? 11. Реакция между кислотой и основанием нротекает по схеме
При ноддержаиии ностоянного значения кислотности в ходе каждого кинетического эксперимента получена следующая зависимость константы скорости от pH:
Найдите константы кислотности 12. Выведите кинетическое уравнение реакции гидрирования олефнна при катализе комплексом 13. Выведите кинетическое уравнение реакции 14. Реакция, катализируемая комплексом металла, протекает по схеме
Выведите кинетическое уравнение и предложите метод его экспериментальной проверки. 1. Реакция лексом
ГЛАВА IV РАДИКАЛЬНЫЕ РЕАКЦИИ Радикальные реакции, протекающие через промежуточное образование свободных атомов и радикалов, занимают важное место в промышленности основного органического и нефтехимического синтеза. К ним принадлежат многие реакции хлорирования, окисления, теломеризации и полимеризации, пиролиз и др. Большинство .радикальных реакций являются цепными и включают три основные стадии: зарождение цепи, при котором образуются свободные радикалы или атомы; продолжение цепи, ведущее к образованию продуктов реакции; обрыв цепи, когда свободные радикалы исчезают. ЗАРОЖДЕНИЕ ЦЕПИ Первоначальное образование свободных радикалов или атомов происходит обычно при гемолитическом разрыве какой-либо связи в молекуле:
Данные, приведенные в табл. 15, показывают, что энергии связей часто достаточно велики, и для разрыва связей необходим подвод энергии извне в виде тепла или излучения. В органических соединениях гомолитический разрыв связи облегчается, когда образующиеся радикалы способны стабилизоваться за счет эффектов сопряжения и сверхсопряжения с не- Таблаца 15. Энергия разрыва Q химических связей
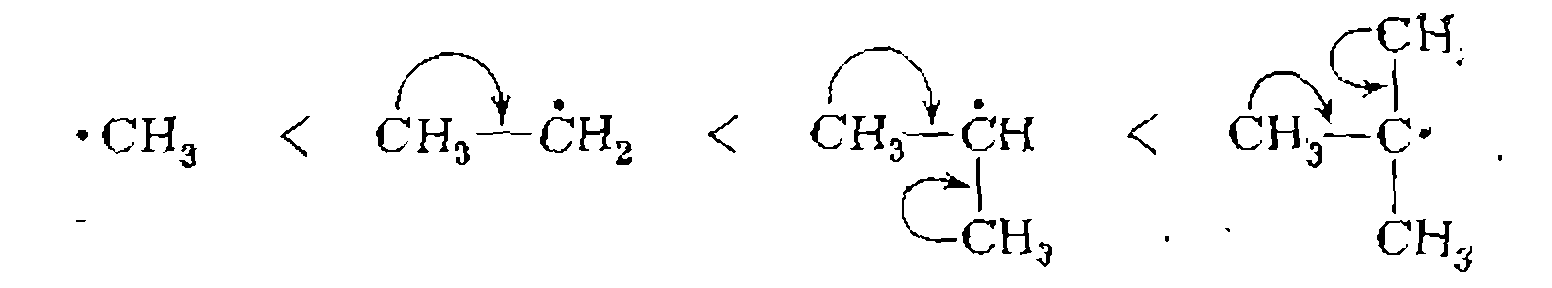
спаренным электроном радикала. Так, стабильность алкильных радикалов и скорость разрыва соответствующих связей меняются в ряду:
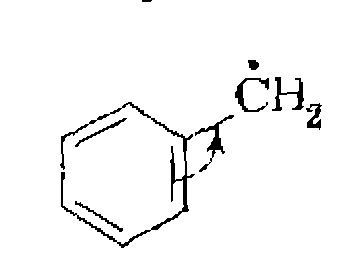
Бензильный и особенно трифени л метальный радикалы стабилизуются за счет сопряжения с ароматической системой связей:
Более стабильные радикалы в то же время менее реакционно- слособиы. Так, трифенилметил настолько ста билен, что степень диссоциации гексафенилэтана даже при комнатной температуре достигает 2%. В большинстве же случаев высокая реакционная способность атомов и радикалов обусловливает очень низкую их концентрацию в реакционных смесях—порядка
Термическое зарождение цепи. В этом случае энергия, необходимая для разрыва связей в молекуле одного из реагентов,, подводится в виде тепла; соответствующий способ проведения реакции называют термическим (термическое окисление, хлорирование, крекинг). Поскольку обратная гомолизу реакция рекомбинации свободных атомов и радикалов происходит при каждом столкновении и имеет нулевую энергию активации, при гомолитическом: расщеплении молекулы энергия активации равна тепловому эффекту, т. е. энергии -разрыва 'Связи: позволяет представить константу скорости таким уравнением:
Зная порядок величины А для мономолекулярных реакций в газовой фазе Из табл. 15 видно, что в молекулах углеводородов наименее- прочной является С—С-связь, и при гомолизе этих молекул вл первую очередь рвется она:
В случае хлорпроизводных зарождение цепи происходит в результате разрыва связи
Гомолитическому разрыву связей нередко способствуют факторы, ведущие к снижению энергии активации за счет одновременного выделения энергии при взаимодействии образующегося радикала с другим веществом. Такое влияние оказывает, например, стенка сосуда или иные твердые поверхности, которые способны адсорбировать второй радикал и о-существлять гетерогенное зарождение цепи:
При реакциях термического окисления такую же роль играет образование связи атома водорода с кислородом:
При галогенировании снижение энергии активации достигается за счет следующих реакций зарождения цепи:
В последнем случае тепловой эффект становится положительным, что обусловливает спонтанное протекание процессов фторирования даже при отрицательных температурах. В большинстве же случаев термические реакции осуществляют при 150— 800 °С. В дальнейшем мы будем обозначать скорость зарождения цепн через
и при бимолекулярных реакциях зарождения цепи:
„ Химическое инициирование цепи. Инициаторами называют вещества, способные расщепляться с образованием реакционно- способных радикалов или атомов при более низкой температуре, чем сами реагенты. Очевидно, что, вводя инициаторы в реакционную массу, можно существенно ускорить зарождение це- пи и провести процесс при более низкой температуре. Это имеет большое значение для интенсификации процесса илн для процессов, в которых исходные вещества либо продукты терми- чески нестабильны. При высокотемпературных реакциях инициатором иногда служит хлор, энергия связи в молекуле которого не столь велика. Такую же роль могут выполнять азотная кислота или ок« сиды азота, неспаренные электроны которых обусловливают инициирование реакции:
В большинстве же случаев инициаторами являются .вещ&ства, малая энергия диссоциации которых на активные радикалы, способные к дальнейшему продолжению цепи, сообщается за счет одновременного образования стабильных молекул — например, азота из азобисдиизобутиронитрила:
диоксида углерода из бензоилпероксида:
ацетона из грег-бутилпероксида:
Инициаторами могут служить также органические перокси- кнслоты ческие пероксидные соединения (персульфаты, пербораты и др.). Все инициаторы распадаются с достаточной скоростью в определенной области температур, например: азобисизобутиронит- рил при 80—90 °С, бензоилпероксид при 90—ЮОХ, грег-бутил- пероксид при 150 X. Очевидно, что те же температуры нужно использовать при проведении реакций, инициируемых этими соединениями. При зарождении цепи в присутствии инициаторов
Более медленной является первая стадия, поэтому скорость зарождения цепи при помощи инициаторов определяется скоростью их распада. Последняя реакция обычно мономолекулярна и описывается таким кинетическим уравнением:
Не все образующиеся радикалы принимают участие в последующем инициировании цепи: часть их гибнет уже в момент распада молекулы инициатора, и, кроме того, наблюдается разложение инициатора под действием других радикалов, ие приво дящее к образованию новых радикалов (так называемая передача цепи через инициатор):
В результате только часть инициатора расходуется полезно, что учитывают введением коэффициента его эффективности
Коэффициент Мономолекулярность расщепления молекул инициаторов строго соблюдается для азобисизобутиронитрила, константа скорости которого не зависит ни от растворителей, ни от концентрации:
У пероксидных соединений первый порядок сохраняется только при низких концентрациях (^0,02 моль/л); повышение концентрации ведет к развитию цепного расщепления. Для этих веществ распад сильно ускоряется солями металлов переменной валентности (Со, Мл, Fe, Си). В их присутствии в инициирующей системе идут окислительно-восстановительные реакции, существеиио изменяющие энергетику процесса:
В отличие от катализаторов инициаторы во время реакции расходуются, и их концентрация постепенно уменьшается. Образующиеся при распаде частицы превращаются затем в стабильные вещества, которые содержатся в продуктах реакции в виде примесей. Другим источником радикалов при химическом инициировании являются окислительно-восстановительные реакции солей и комплексов металлов переменной валентности с некоторыми исходными реагентами:
Реакции фотолиза и радиолиза. При облучении УФ-светом или другими видами излучений (рентгеновским, При фотохимическом инициировании молекула разрывается по самой слабой связи (см. табл. 15) с образованием двух радикалов:
Для разрыва связи энергия поглощаемого кванта
Из этого соотношения можно подсчитать, что для фотолиза молекулы хлора необходим свет длиной волны 494 нм (сине-зеленый), для разрыва С—Н-связи нужен свет 268 нм (УФ-об- ласть) и т. д. При фотохимическом процессе иногда используют сенсибилизаторы, роль которых состоит в передаче энергии, поглощенной ими, реагирующим молекулам. Так, атомы ртути способны легко переходить в возбужденное состояние и отдавать поглощенный ими квант:
Применение сенсибилизаторов оправдывается, если реагенты слабо поглощают свет в приемлемой области спектра. Под влиянием жесткого рентгеновского излучения или
Инициирующее действие излучения зависит от его интенсивности I, равной потоку квантов через единицу поперечного сечения за единицу времени. Этот поток при прохождении через реакционную массу уменьшается в соответствии с уравнением Ламберта — Бера:
где степень поглощения света мала, интенсивность поглощенного излучения в расчете на единицу объема за единицу времени составит
В фотохимии она измеряется в Эйнштейнах (Э)* на 1 л в секунду. Аналогичные зависимости наблюдаются и при радиационнохимических процессах, где мощность поглощенной дозы Р измеряется в рентгенах за единицу времени. Не все поглощенное излучение расходуется полезно на зарождение цепи; часть его тратится на фосфоресценцию, люминесценцию, переходит в тепловую энергию и т. д. Кроме того, какое-то количество радикалов немедленно рекомбинирует или диспропорционирует, не участвуя в зарождении цепи. Если обозначить через
Если же излучение поглощается полностью, скорость зарождения цепи не зависит от концентрации
и особенностью такого процесса является изменение скорости инициирования по объему, что связано с постепенным снижением интенсивности по мере удаления от источника облучения. ПРОДОЛЖЕНИЕ И ОБРЫВ ЦЕПИ Образовавшийся при зарождении цепи свободный атом или радикал, обладая высокой реакционной способностью, начинает цепь превращений, ведущих к образованию продуктов реакции. Эта цепь складывается из элементарных стадий радикального замещения, расщепления или присоединения, специфичных для каждой конкретной реакции. Они подробно рассмотрены ниже; здесь же можно ограничиться следующей схемой:
Во второй реакции образовался тот же свободный радикал
Совокупность элементарных стадий (например, написанная выше), в результате которой образуется радикал, начинавший цепь, называется ее звеном. Число повторяющихся звеньев, составляющих цепь, называют ее длиной. В разных реакциях длина цепи меняется от нескольких единиц илн десятков до mho- гих тысяч. По этой причине уже небольшое количество инициатора илл поглощенных при облучении квантов энергии ведет к образованию большого количества продуктов, что является одним из доказательств цепного механизма процесса. При использовании инициаторов, имеющих коэффициент
где коэффициент п равен числу молекул продукта, образующихся в каждом звене цепи (обычно п = 1), При фотохимических реакциях цепной процесс характеризуется квантовым выходом
Радиационно-химические реакции ( зуются числом прореагировавших молекул на 100 эВ поглощенного излучения (радиационно-химический выход G). Эта величина связана с длиной цепи более сложной зависимостью, однако для типичного цепного процесса обычно G>3. Если длина цепи достаточно велика (^10), в стационарных условиях реакции скорости всех элементарных стадий продолжения цепи равны. Из этого условия имеем:
Таким образом, соотношение стационарных концентраций разных свободных радикалов обратно пропорционально произведению констант скоростей элементарных стадий и концентра-' ций реагентов, с которыми эти радикалы взаимодействуют. Часто бывает, что одно из произведений много меньше другого, и тогда соответствующую стадию продолжения цепи называют лимитирующей. В таком случае в реакционной смеси преобладает тот радикал, который участвует в лимитирующей стадии. Он обычно является наименее реакционноспособным среди других радикалов, присутствующих в реакционной массе. Обрыв цепи ведет к исчезновению свободного радикала нли по крайней мере к превращению его в более стабильный радикал, не способный к продолжению цепи. По отношению к радикалам реакции обрыва могут быть моно- или бимолекулярными, в соответствии с чем различают квадратичный и линейный обрывы цепи. При квадратичном обрыве цепи в результате бимолекулярного взаимодействия двух радикалов их свободные электроны взаимно спариваются и образуются молекулярные продукты: 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|
 К ним относятся катиониты сульфопо-
К ним относятся катиониты сульфопо-

 на полистирольной основе (АВ-17), а также аминогруппы, например
на полистирольной основе (АВ-17), а также аминогруппы, например  в поливинил-
в поливинил-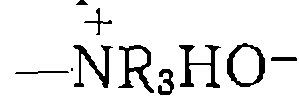 , обладают меньшей термостойкостью, чем катиониты, и их применение ограничивается реакциями, протекающими при температурах ниже 60—70 °С. Они эффективно катализируют, в частности, реакцию присоединения воды к акрилонитрилу при нуклеофильном катализе гидроксил-ионом:
, обладают меньшей термостойкостью, чем катиониты, и их применение ограничивается реакциями, протекающими при температурах ниже 60—70 °С. Они эффективно катализируют, в частности, реакцию присоединения воды к акрилонитрилу при нуклеофильном катализе гидроксил-ионом: 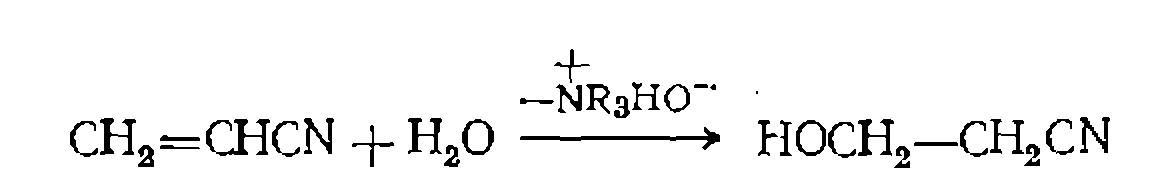
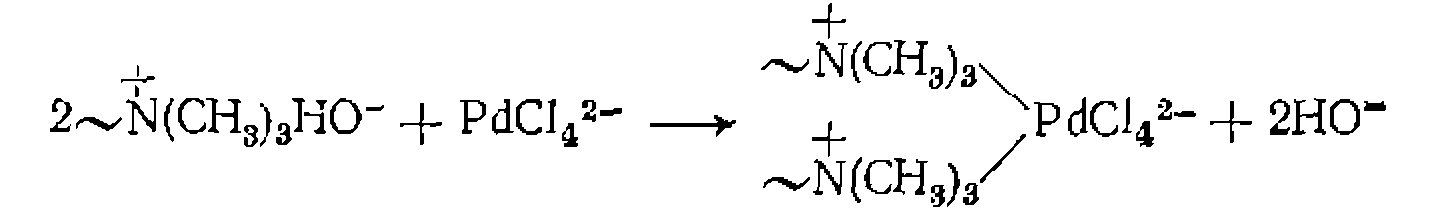
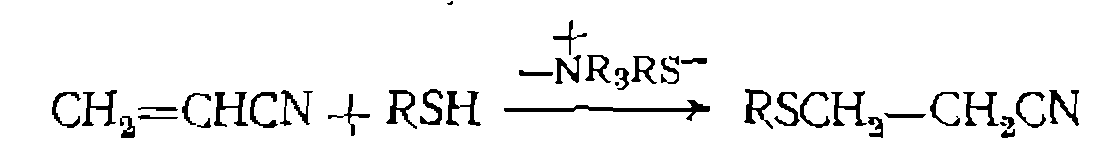
 на анионите АВ-17 приводит к образованию активного катализатора гидрирования олефинов:
на анионите АВ-17 приводит к образованию активного катализатора гидрирования олефинов: иммобилизуется путем взаимодействия с
иммобилизуется путем взаимодействия с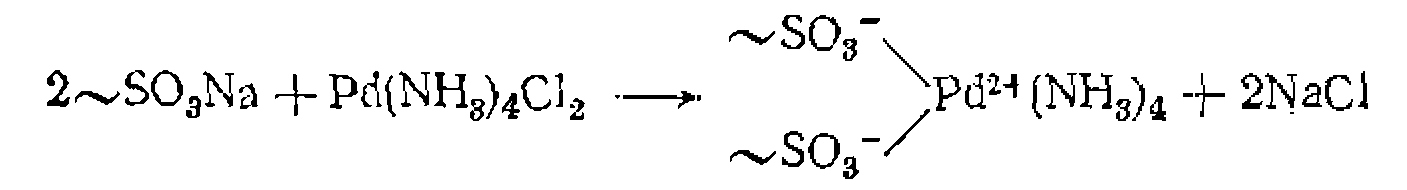
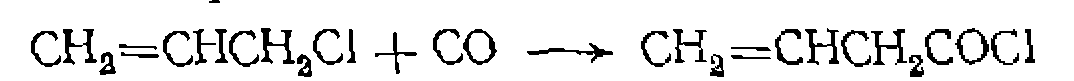
 или
или , поливинилхлорид, -в котором хлор замещен на группы .
, поливинилхлорид, -в котором хлор замещен на группы .  . поливиниловыйспирт, обработанный диалкил (арил)хлорфосфинами
. поливиниловыйспирт, обработанный диалкил (арил)хлорфосфинами  , аниониты
, аниониты и т. л. Гетерогенизация комплек
и т. л. Гетерогенизация комплек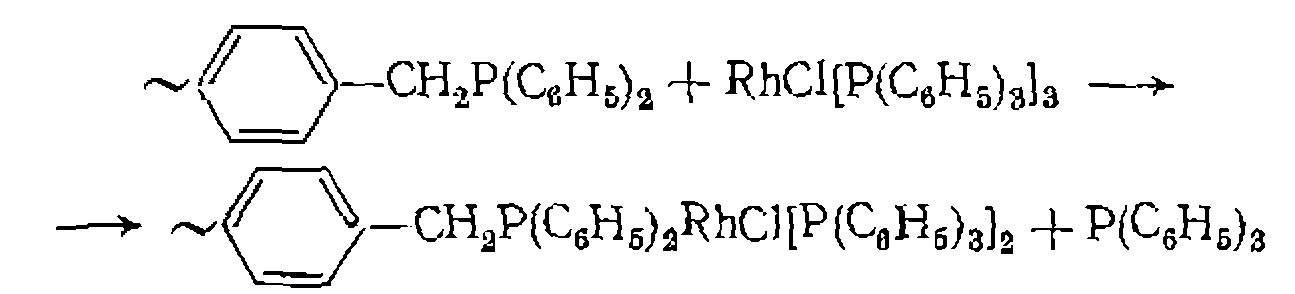
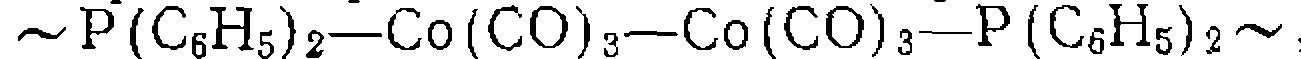
 олигомеризации олефинов
олигомеризации олефинов  и т. п.
и т. п.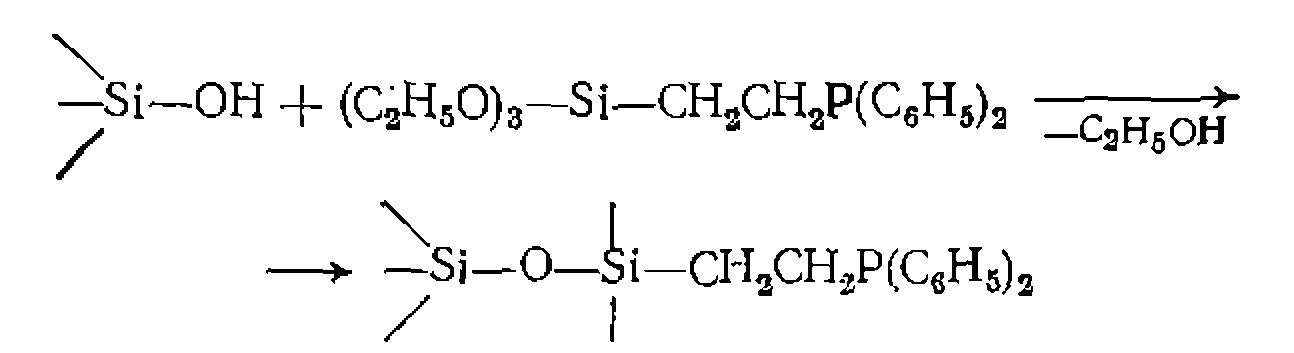
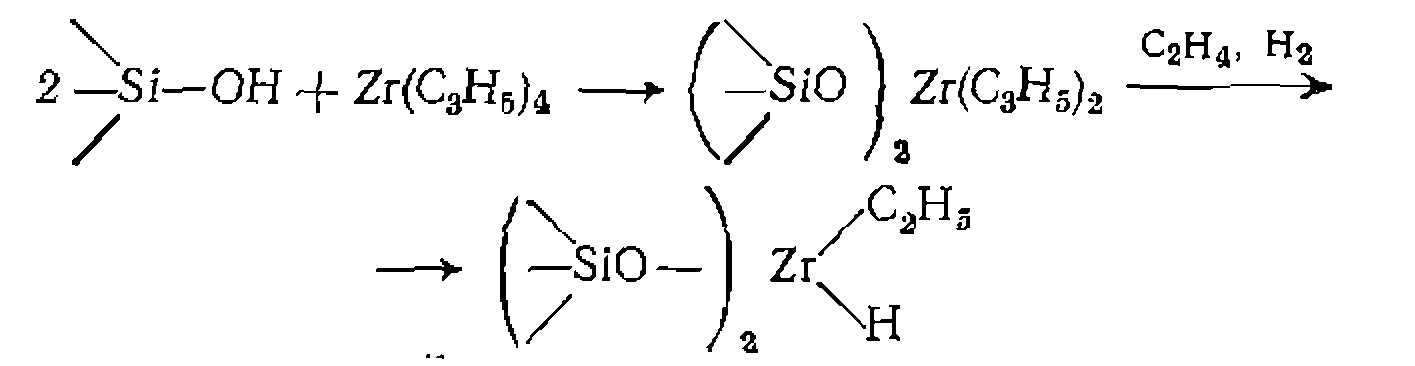
 и
и с силикагелем, алюмосиликатами и оксидом алюминия.
с силикагелем, алюмосиликатами и оксидом алюминия.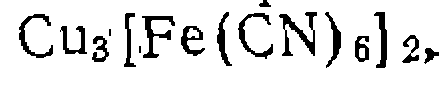
 в гликоле добавляют к суспензии
в гликоле добавляют к суспензии  в метаноле и после ис
в метаноле и после ис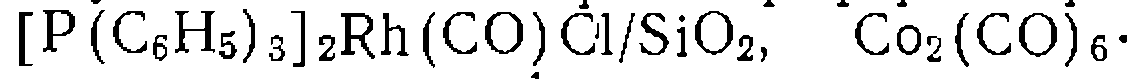
 , активные как в газофазном, так и в жидкофазном процессе. Пропиткой пористых минеральных носителей кислотами получают гетерогенизированные кислотные катализаторы, используемые преимущественно в газофазных процессах дегидратации и этерификации (например,
, активные как в газофазном, так и в жидкофазном процессе. Пропиткой пористых минеральных носителей кислотами получают гетерогенизированные кислотные катализаторы, используемые преимущественно в газофазных процессах дегидратации и этерификации (например,  на
на  и т. п.).
и т. п.). н
н 
 »-группой) и гидрирование продуктов конденсации до 2-этилгексаналя (катализатор Rh).
»-группой) и гидрирование продуктов конденсации до 2-этилгексаналя (катализатор Rh). катализируемой
катализируемой  выведите кинетическое уравнение, если известно, что параллельно нротекает иекаталитическая реакция, а катализ осуществляется по схеме
выведите кинетическое уравнение, если известно, что параллельно нротекает иекаталитическая реакция, а катализ осуществляется по схеме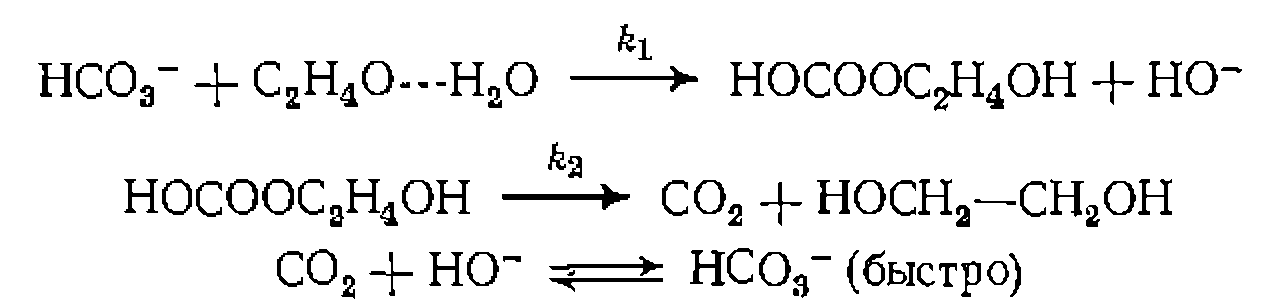
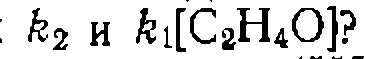
 начальная скорость реакции
начальная скорость реакции  пропорциональна
пропорциональна  и не зависит от
и не зависит от  В то же время зависимость “
В то же время зависимость “  от t описывается уравнением
от t описывается уравнением  зависит от
зависит от  ,. Каким уравнением описываются все полученные экспериментальные данные и как
,. Каким уравнением описываются все полученные экспериментальные данные и как  зависит от
зависит от 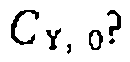
 при катализе
при катализе  нротекает по механизму
нротекает по механизму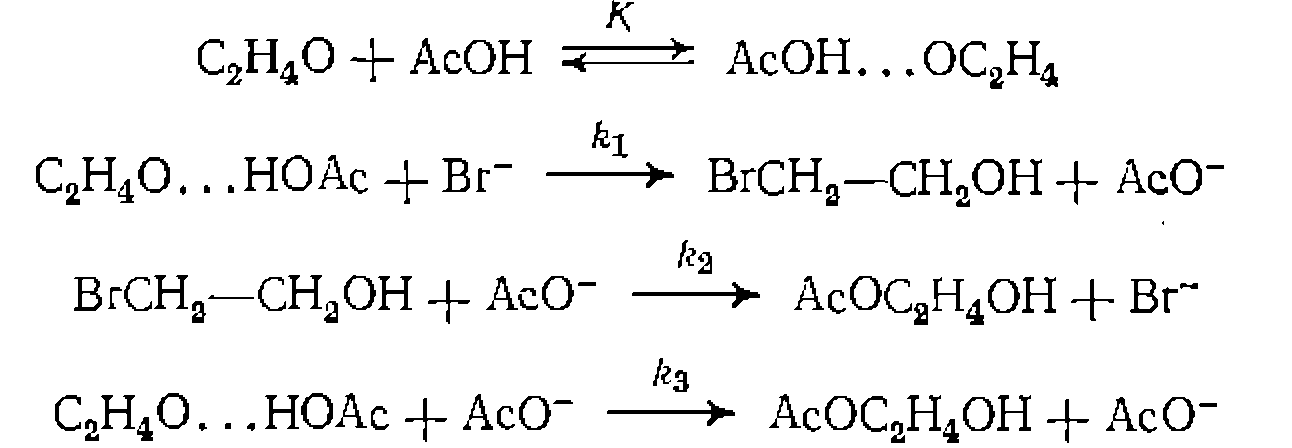

 гидроксиэтил) пиридин и описывается кинетическим уравнением
гидроксиэтил) пиридин и описывается кинетическим уравнением  . Какому механизму соответствует эта кинетика?
. Какому механизму соответствует эта кинетика?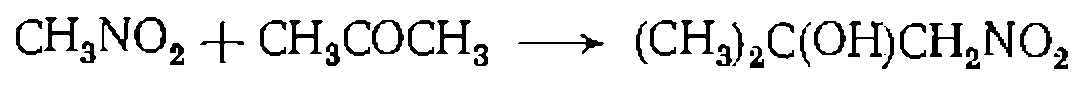
 вещества А, константа скорости нревращения которого в В при разных значениях функций кислотности
вещества А, константа скорости нревращения которого в В при разных значениях функций кислотности  равна:
равна: 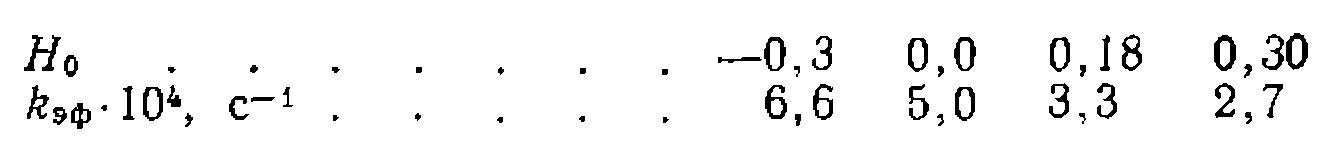
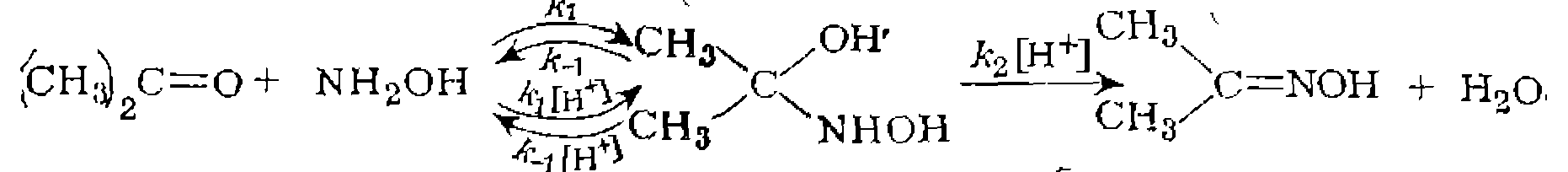
 в зависимости от
в зависимости от  со значениями кинетических констант и константы кислотности
со значениями кинетических констант и константы кислотности 
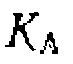 веществ АН и ВН+ н константу скорости k[.
веществ АН и ВН+ н константу скорости k[. механизм которой приведем на с. 194.
механизм которой приведем на с. 194. с этиленом, механизм которой приведен на с. 198, нрн условии, что концентрации всех промежуточных комнлексов малы но сравнению с
с этиленом, механизм которой приведен на с. 198, нрн условии, что концентрации всех промежуточных комнлексов малы но сравнению с 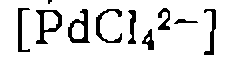
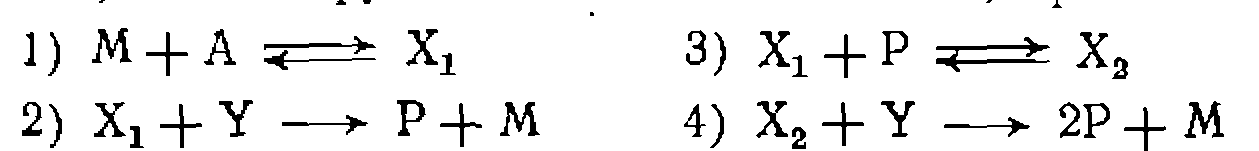
 катализируется комп
катализируется комп и описывается кинетическим уравнением
и описывается кинетическим уравнением . Каков ее механизм?
. Каков ее механизм?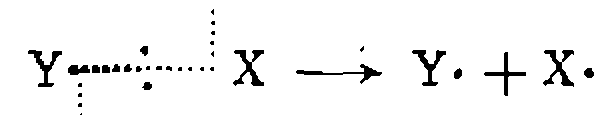
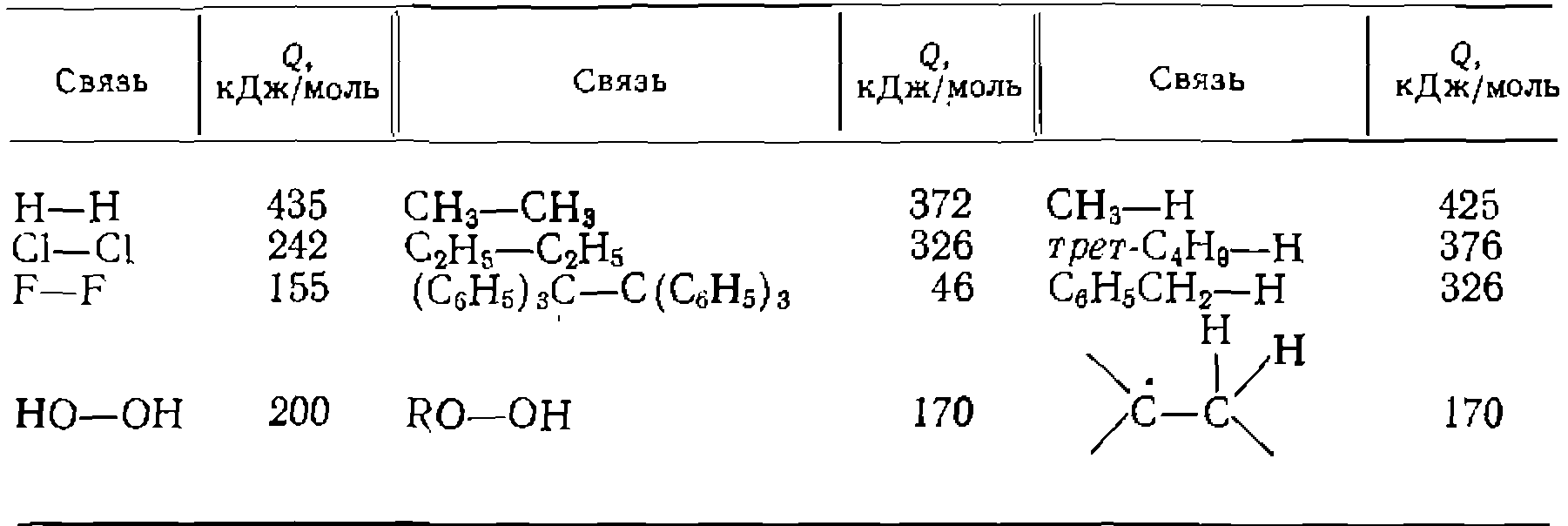
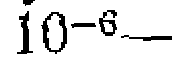
 моль/л. Поэтому идентификация их достижима лишь по результатам дальнейших превращений или с помощью спектров электронного парамагнитного резонанса (ЭПР).
моль/л. Поэтому идентификация их достижима лишь по результатам дальнейших превращений или с помощью спектров электронного парамагнитного резонанса (ЭПР). . Это
. Это (ПМ)
(ПМ) и анергию разрываемой связи, можно- найти область температур, в которой гомолиз будет протекать с достаточно высокой скоростью. Так, чтобы время полураспада составляло около 2 ч
и анергию разрываемой связи, можно- найти область температур, в которой гомолиз будет протекать с достаточно высокой скоростью. Так, чтобы время полураспада составляло около 2 ч  5 для расщепления молекулы С12 требуется температура 480 для разрыва С—Освязи — около 700 °С, и т. д.
5 для расщепления молекулы С12 требуется температура 480 для разрыва С—Освязи — около 700 °С, и т. д.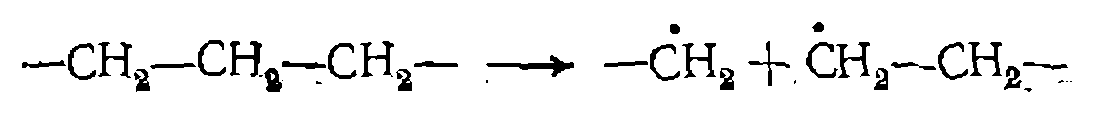
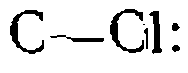


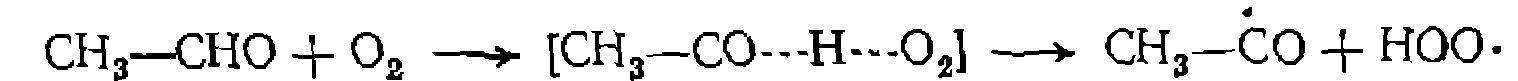
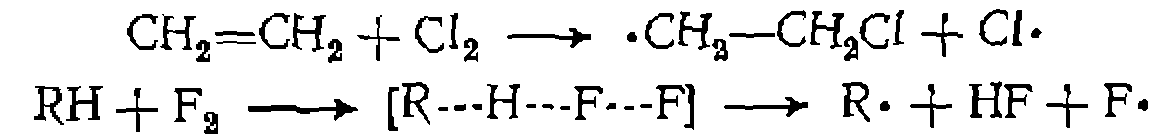
 Тогда имеем при момомолекулярном распаде
Тогда имеем при момомолекулярном распаде (1V-2)
(1V-2) (IV-3)
(IV-3)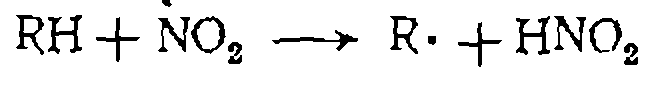
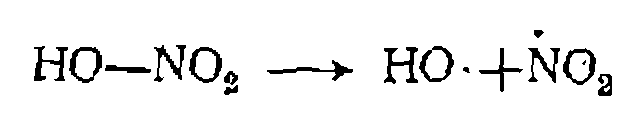

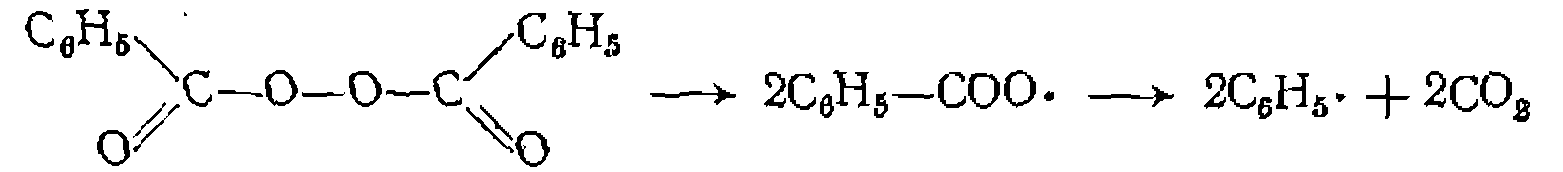
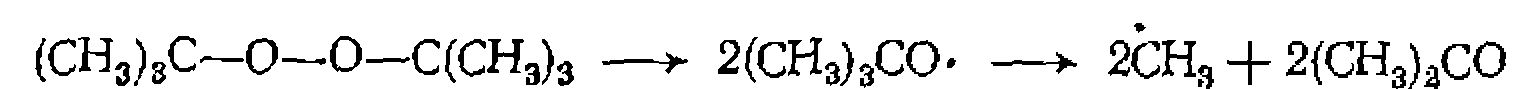
 , гидропероксиды
, гидропероксиды  водород и неоргани
водород и неоргани вначале образуется специфический для каждого из них радикал, который быстро взаимодействует с одним из реагентов и дает радикал, ведущий дальнейшую цепь, например:
вначале образуется специфический для каждого из них радикал, который быстро взаимодействует с одним из реагентов и дает радикал, ведущий дальнейшую цепь, например: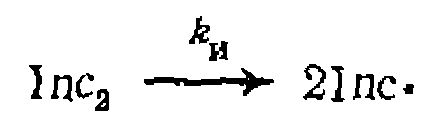
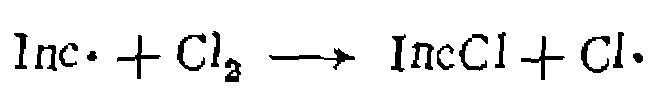
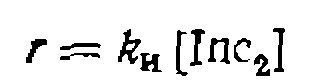 (IV-4)
(IV-4)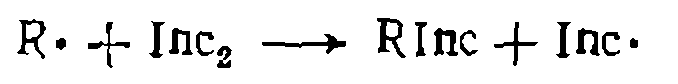
 при этом скорость зарождения цепи с инициаторами выражается кинетическим уравнением:
при этом скорость зарождения цепи с инициаторами выражается кинетическим уравнением: (IV-5)
(IV-5) близок к единице для бензоилпероксида, но достигает лишь ?^0,6 для азобисизобутиронитрила и составляет еще меньшую величину для некоторых гидропероксидов.
близок к единице для бензоилпероксида, но достигает лишь ?^0,6 для азобисизобутиронитрила и составляет еще меньшую величину для некоторых гидропероксидов.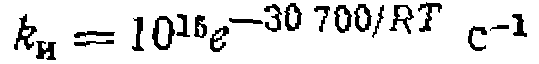
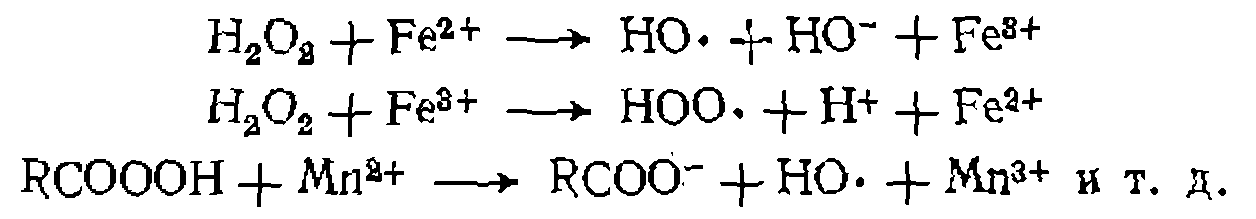
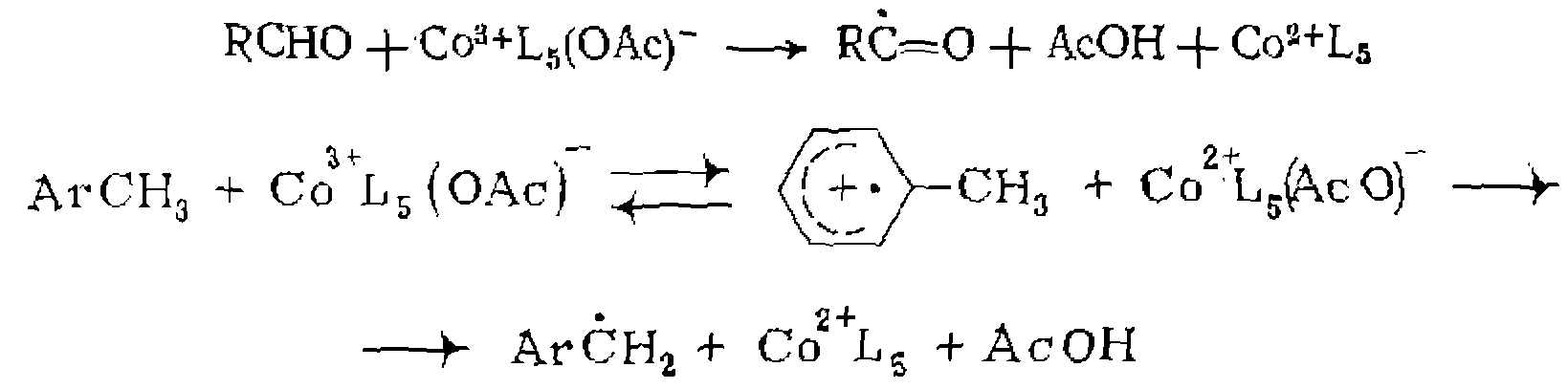
 лучами) молекулы под влиянием поглощенных ими квантов энергии переходят в возбужденное состояние. При достаточно большой энергии кванта происходит разрыв связей и образование свободных радикалов. Поскольку возбуждение молекулы происходит под влиянием не температуры, а внешнего источника энергии, скорость распада вообще не зависит от температуры, а определяется интенсивностью облучения. В этом состоит большое преимущество фото- и радиационно-химических способов проведения реакций, вполне осуществимых при низких температурах.
лучами) молекулы под влиянием поглощенных ими квантов энергии переходят в возбужденное состояние. При достаточно большой энергии кванта происходит разрыв связей и образование свободных радикалов. Поскольку возбуждение молекулы происходит под влиянием не температуры, а внешнего источника энергии, скорость распада вообще не зависит от температуры, а определяется интенсивностью облучения. В этом состоит большое преимущество фото- и радиационно-химических способов проведения реакций, вполне осуществимых при низких температурах.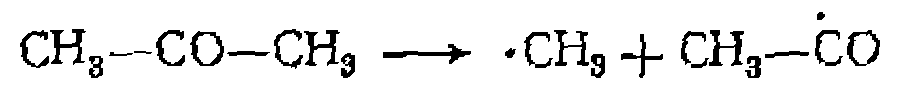
 не может быть ниже энергии связи, что дает соотношение
не может быть ниже энергии связи, что дает соотношение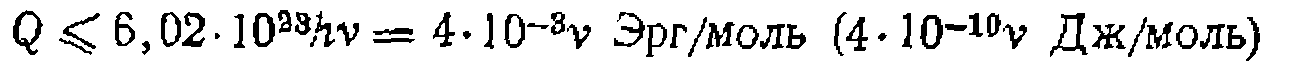
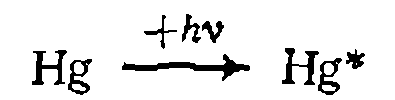
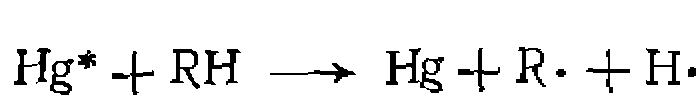
 излучения с большой энергией кванта, значительно превосходящей энергию связи, способны разрываться все связи в молекуле с образованием не только радикалов, но и ионов. Однако преимущественно рвутся те связи, которых больше в единице объема, или же связи, занимающие в молекуле периферийное положение, напр'кмер С—Н в молекуле углеводородов:
излучения с большой энергией кванта, значительно превосходящей энергию связи, способны разрываться все связи в молекуле с образованием не только радикалов, но и ионов. Однако преимущественно рвутся те связи, которых больше в единице объема, или же связи, занимающие в молекуле периферийное положение, напр'кмер С—Н в молекуле углеводородов:
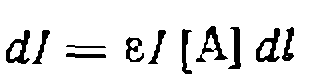 (IV-6)
(IV-6) —коэффициент поглощения; [А]—концентрация вещества, поглощающего свет; I — толщина поглощающего слоя. Если
—коэффициент поглощения; [А]—концентрация вещества, поглощающего свет; I — толщина поглощающего слоя. Если {IV* 7)
{IV* 7)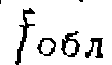 коэффициент, учитывающий долю полезно израсходованного излучения, скорость зарождения цепи при малой степени поглощения будет равна
коэффициент, учитывающий долю полезно израсходованного излучения, скорость зарождения цепи при малой степени поглощения будет равна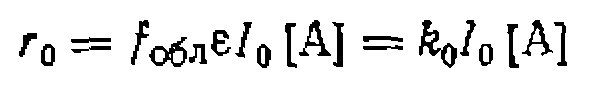 (IV-8)
(IV-8) или
или  (IV-9)
(IV-9)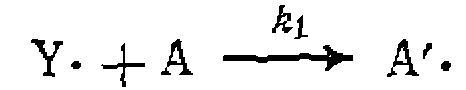
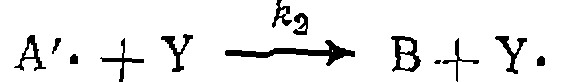
 с которого началось превращение. Благодаря этому последовательность элементарных реакций может повториться вновь, затем еще и еще, пока свободный ради кал не исчезнет по той или иной причине. Таким образом и осуществляется цепной процесс, в котором один радикал, образовавшийся на стадии зарождения цепи, приводит к образованию многих молекул продукта В.
с которого началось превращение. Благодаря этому последовательность элементарных реакций может повториться вновь, затем еще и еще, пока свободный ради кал не исчезнет по той или иной причине. Таким образом и осуществляется цепной процесс, в котором один радикал, образовавшийся на стадии зарождения цепи, приводит к образованию многих молекул продукта В.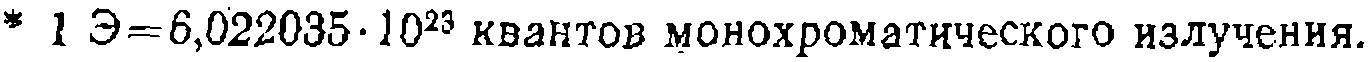
 близкий к 1 (бензоилпероксид), среднюю длину цепи v можно определить по количеству образовавшегося продукта ГВ1 и по количеству израсходованного инициатора
близкий к 1 (бензоилпероксид), среднюю длину цепи v можно определить по количеству образовавшегося продукта ГВ1 и по количеству израсходованного инициатора 
 (IV-10)
(IV-10) , который равен числу прореагировавших молекул на один поглощенный квант света:
, который равен числу прореагировавших молекул на один поглощенный квант света: (IV-11)
(IV-11) ) характери
) характери (IV-12)
(IV-12)