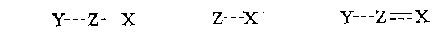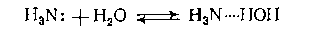|
|
МЕХАНИЗМ И КИНЕТИКА ЭЛЕМЕНТАРНЫХ РЕАКЦИЯПосле выдвижения гипотезы о схеме превращений наступает следующий этап обработки опытов — выдвижение гипотезы о механизме каждой простой реакции или об их совокупности и построение кинетических уравнений. Известно, что с точки зрения механизма все реакции делятся наэлементарные и неэлементарные. Первые протекают в одну необратимую стадию без образования каких-либо промежуточных частиц или комплексов (к ним не относятся переходное состояние или активированный комплекс, через которые идег любая элементарная реакция). К элементарным реакциям полностью применим закон действующих масс, и их скорость пропорциональна концентрации каждого реагента в степени, равной его стехиометрическому коэффициенту. По числу молекул, принимающих участие в элементарном акте, такие реакции бывают моно-, би- н (очень редко) тримолекулярными
где [А] или [Y] —концентрации или парциальные давления веществ, ak— константы скорости. Известно, что порядок реакции по какому-либо веществу' равен показателю степени, в которой его концентрация входит в кинетическое уравнение, а суммарный порядок равен сумме этих показателей. Нетрудно видеть, что для элементарных реакций порядок, молекулярность и стехиометрические коэффициенты совпадают. В зависимости от порядка реакции размерность констант скорости различна и может быть найдена делением размерности скорости на размерность произведения концентраций или парциальных давлений. Так, для гомогенных жидкофазных реакций размерность констант будет такой:
Для жидкофазной гетерогенно-каталитической реакции получим такое общее выражение для размерности константы скорости:
Для газофазных реакций вместо концентраций часто употребляют парциальные давления, поэтому размерность констант будет такова:
Таковы же размерности констант и для соответствующих неэлементарных реакций.
Напомним некоторые положения теории элементарных реакций. Известно, что каждая из них протекает через переходное состояние или активированный комплекс, в котором произошло частичное образование новых химических связей и ослабление прежних. Для элементарных реакций замещения, расщепления и присоединения принято эти переходные состояния изображать так:
между ней и энергией начального состояния равна тому энергетическому барьеру, который должны преодолеть реагенты. Эта разность и есть энергия активации элементарной реакции, а разность между начальной и конечной энергией равна энтальпии реакции АН. Очевидно, что для обратимых реакций существует соотношение:
В теории абсолютных скоростей реакций Эйринг и Поляни приняли, что активированный комплекс находится в обычном термодинамическом равновесии с исходными реагентами. Это равновесие можно охарактеризовать активационными параметрами; свободной энергией активации энтропией активации и уравнения Аррениуса приводит к такому выражению для константы скорости элементарной реакции:
Из его сравнения с обычной формой уравнения Аррениуса (к = = Koe-E/RT)видно, что предэкспоненциальный множитель последнего является функцией энтропии активации. Энтропию "активации можно найти из экспериментальных данных по кинетике, имея в виду, что ek/h=5,662* 10-10 с-1, а предэкспоненциальный множитель k0 найден для концентраций, выраженных в моль/л. Энтропия активации дает некоторое представление о механизме элементарных реакций, так как она связана с изменением упорядоченности системы при образовании активированного комплекса. Для бимолекулярных реакций эта упорядоченность возрастает, а энтропия активации имеет отрицательное значение. Наоборот, для мономолекулярных реакций переходное состояние из-за удлинения рвущихся связей становится менее упорядоченным, и энтропий активации приобретает положительное значение.
Рис. II. Влияние диэлектрической проницаемости растворителя на скорость реакции между ионом и молекулой (о) и между двумя полярными молекулами (5). Рассмотренные представления наиболее важны для элементарных реакций в газовой фазе. Для жидкофазных реакций картина существенно усложняется из-за взаимодействий реагентов и активированного комплекса со средой, объединяемых понятием сольватации. Последняя может быть неспецифической, обусловленной главным образом электростатическим взаимодействием реагентов и активированного комплекса с растворителем, и специфической, зависящей от возникновения химических связей реагентов н переходного комплекса со средой.
Существуют методы количественного описания влияния среды на скорость химических реакций. При электростатической сольватации принимают, что растворитель является непрерывной однородной средой. Исходя из этого и пз теории абсолютных скоростей реакций для взаимодействия между двумя ионами или ионом и диполем были обоснованы соответственно уравнения Скетчарда и Амиса, которые дают линейную связь логарифма константы скорости с обратным значением диэлектрической проницаемости среды: Эти уравнения -соответствуют линейной зависимости lnkот 1/sили от (е—1)/(2e+l) (рис. II). Зависимости, даваемые уравнениями (11-33) и (II-34), а также приведенные на рис. 11, наглядно интерпретируются следующим образом. При взаимодействии между двумя ионами
В результате комплекс становится менее способным к электростатической сольватации, чем исходные реагенты, величина Gувеличивается при переходе к более полярным растворителям, а константа скорости уменьшается. В противоположность этому при реакции двух нейтральных молекул
переходное состояние более полярно и сильнее сольватируется, чем исходные реагенты, и при повышении диэлектрической постоянной среды константа скорости увеличивается. Специфическая сольватация нередко проявляется сильнее, чем электростатическая, и бывает обусловлена главным образом кислотно-основным взаимодействием реагентов с растворителем. Так, положительно заряженные ионы обычно сольватированы основаниями (вода, спирты), а анионы — молекулами растворителя, обладающего кислотными свойствами и способного к образованию водородных связей:
Такие растворители в меньшей степени сольватируют и нейтральные молекулы, содержащие атомы с неподеденными парами электронов:
Результат сольватации реагентов может быть разным. Когда во время реакции в данном реагенте происходит разрыв каких-либо связей, то его специфическая сольватация может ускорять такую реакцию:
В других случаях специфическая сольватация снижает активность реагента из-за рассредоточения заряда или уменьшения электронной плотности на реакционном центре. Этот эффект особенно сказывается на реагентах-анионах, и в результате, например, реакция
из-за сольватации бромид-иона метанолом протекает в этом растворителе в 58 000 раз медленнее, чем в диметилформамиде, имеющем ту же диэлектрическую проницаемость. Элементарными являются лишь немногие из органических реакций, например нуклеофильные процессы, протекающие по 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|