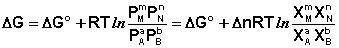|
|
Примеры термодинамических расчетовЗадача1. Возможен ли при стандартных условиях процесс взаимодействия азота и кислорода воздуха? Решение. Значение DG (N2) + (O2) = 2(NO) DH S DG 1 способ. Рассчитываем DН DН DS DG 2 способ. Изменение энергии Гиббса в ходе реакции можно также рассчитать, зная величины изменения энергии Гиббса образования веществ, участвующих в химическом процессе, по формуле DG Полученное значение практически совпадает с рассчитанным первым способом. Так как DG
Взаимосвязь стандартной энергии Гиббса и константы равновесия выражается уравнением DG Пример 2. Определите давление разложения 3[Fe2O3] ⇄ ⇄ 2[Fe3O4] + ½(O2) при 627оС. При какой температуре давление кислорода достигнет 1 атм (101 325 Па)? Решение. Для определения давления разложения, т. е. давления кислорода, который образуется в данном процессе термического разложения, надо вычислить Кр = р Запишем процесс и для всех веществ, участвующих в реакции, выписываем из справочника значения DH 3[Fe2O3] ⇄ 2[Fe3O4] + ½(O2) DH S В соответствии со следствием из закона Гесса: DН DS Изменение стандартной энергии Гиббса при 900 К находим из соотношения: DН DG lgKp = Для данного гетерогенного процесса Кр = р Р(О Если давление кислорода равно 1 атм то lgKp = lg1 Найдем температуру, при которой DG DG Т = Следовательно, при температуре 1686 К, когда давление кислорода равно внешнему (1 атм), начнется разложение Fe2O3. Пример 3. Определить термодинамические характеристики DH Решение. Для процесса {SnBr2} ⇄ (SnBr2) Kр = РSnBr
Kp T1 = 516 + 273 = 789 К, T2 = 909 К. Так как ln1 = 0, то из второго уравнения можно выразить DH –8,314 × 789 × ln0,13 = 909DS DS Тогда DH Полученные значения DH Пример 4. Диссоциация нитрозилхлорида происходит по схеме 2(NOCl) = 2(NO) + (Cl2). Определить, при какой температуре Kр этой реакции равна 1. Чему равно парциальное давление Cl2, если общее давление в системе при равновесии составляет 2 атм? Решение.Запишем уравнение реакции. Для каждого вещества, участвующего в ней, выпишем из справочника значения DH 2(NOCl) = 2(NO) + (Cl2) DH S Рассчитаем DН DН DS Из равенства DН рассчитаем искомую температуру, при которой Kp = 1 (ln1 = 0): Т = Для расчета парциального давления хлора (pCl Кр процесса имеет вид: Кр = Если парциальное давление хлора принять за х, то парциальное давление NO составит 2х, так как из уравнения реакции следует, что в состоянии равновесия на 1 моль хлора образуется 2 моля NO, поэтому парциальные давления компонентов Cl2 и NO тоже будут относиться как 1:2 (pNO = 2pCl Парциальное давление рNOCl можно выразить через парциальные давления pCl рNOCl = Робщ – (pNO + pCl Выражение константы равновесия запишется: Kр = Решая уравнение относительно х, получим х = 0,4595. pCl
ХИМИЧЕСКАЯ КИНЕТИКА. СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ Методы и приемы химической термодинамики позволяют выяснить лишь принципиальную возможность осуществления химической реакции, условия ее вероятного протекания, определить полноту превращения реагирующих веществ в продукты реакции. Законы химической термодинамики позволяют определить направление и предел протекания возможного при данных условиях химического процесса, а также его энергетический эффект. Однако термодинамика не может ответить на вопросы о том, как осуществляется данный процесс и с какой скоростью. Эти вопросы – механизм и скорость химической реакции – и являются предметом химической кинетики. Скорость химической реакции есть число элементарных актов химической реакции, происходящих в единицу времени в единице объема (для гомогенных реакций) или на единице поверхности (для гетерогенных реакций). Скорость химической реакции есть изменение концентрации реагирующих веществ в единицу времени. Первое определение является наиболее строгим; из него следует, что скорость химической реакции можно также выражать как изменение во времени любого параметра состояния системы, зависящего от числа частиц какого-либо реагирующего вещества, отнесенное к единице объема или поверхности – электропроводности, оптической плотности, диэлектрической проницаемости и т.п. Однако наиболее часто в химии рассматривается зависимость концентрации реагентов от времени. В случае односторонних (необратимых) химических реакций очевидно, что концентрации исходных веществ во времени постоянно уменьшаются (ΔСисх < 0), а концентрации продуктов реакции увеличиваются (ΔСпрод > 0). Скорость реакции считается положительной, поэтому математически определение средней скорости реакции в интервале времени Δt записывается следующим образом:
Единицей измерения скорости является [моль/(дм3 с)]. В различных интервалах времени средняя скорость химической реакции имеет разные значения; истинная (мгновенная) скорость реакции определяется как производная от концентрации по времени:
Графическое изображение зависимости концентрации реагентов от времени есть кинетическая кривая (рисунок 1):
Рис. 1.Кинетические кривые для исходных веществ (А) и продуктов реакции (В). Истинную скорость реакции можно определить графически, проведя касательную к кинетической кривой; истинная скорость реакции в данный момент времени равна по абсолютной величине тангенсу угла наклона касательной:
Рис. 2 Графическое определение Vист.
Необходимо отметить, что в том случае, если стехиометрические коэффициенты в уравнении химической реакции неодинаковы, величина скорости реакции будет зависеть от того, изменение концентрации какого реагента определялось. Очевидно, что в реакции 2Н2 + О2 ––> 2Н2О концентрации водорода, кислорода и воды изменяются в различной степени: Молекулярность элементарных реакций Элементарными (простыми) называют реакции, идущие в одну стадию. Их принято классифицировать по молекулярности: Молекулярность элементарной реакции – число частиц, которые, согласно экспериментально установленному механизму реакции, участвуют в элементарном акте химического взаимодействия. Мономолекулярные – реакции, в которых происходит химическое превращение одной молекулы (изомеризация, диссоциация и т. д.): I2 ––> I• + I• Бимолекулярные – реакции, элементарный акт которых осуществляется при столкновении двух частиц (одинаковых или различных): СН3Вr + КОН ––> СН3ОН + КВr Тримолекулярные – реакции, элементарный акт которых осуществляется при столкновении трех частиц: О2 + NО + NО ––> 2NО2 Реакции с молекулярностью более трёх неизвестны. Одной из задач, стоящих перед химической кинетикой, является определение состава реакционной смеси (т.е. концентраций всех реагентов) в любой момент времени, для чего необходимо знать зависимость скорости реакции от концентраций. В общем случае, чем больше концентрации реагирующих веществ, тем больше скорость химической реакции. В основе химической кинетики лежит т. н. основной постулат химической кинетики: Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в некоторых степенях. Для реакции аА + bВ + dD + ... ––> еЕ + ... можно записать:
Коэффициент пропорциональности k есть константа скорости химической реакции. Константа скорости численно равна скорости реакции при концентрациях всех реагирующих веществ, равных 1 моль/л. Зависимость скорости реакции от концентраций реагирующих веществ определяется экспериментально и называется кинетическим уравнением химической реакции. Очевидно, что для того, чтобы записать кинетическое уравнение, необходимо экспериментально определить величину константы скорости и показателей степени при концентрациях реагирующих веществ. Показатель степени при концентрации каждого из реагирующих веществ в кинетическом уравнении химической реакции (в уравнении соответственно x, y и z) есть частный порядок реакции по данному компоненту. Сумма показателей степени в кинетическом уравнении химической реакции (x + y + z) представляет собой общий порядок реакции. Следует подчеркнуть, что порядок реакции определяется только из экспериментальных данных и не связан со стехиометрическими коэффициентами при реагентах в уравнении реакции. Стехиометрическое уравнение реакции представляет собой уравнение материального баланса и никоим образом не может определять характера протекания этой реакции во времени. Для элементарных реакций, проводимых при близких концентрациях исходных веществ, величины молекулярности и порядка реакции совпадают. Тем не менее, никакой чётко определенной взаимосвязи между понятиями молекулярности и порядка реакции не существует, поскольку порядок реакции характеризует кинетическое уравнение реакции, а молекулярность – механизм реакции.
Зависимость скорости химической реакции от температуры Скорость химической реакции значительно зависит от температуры. Не всякое столкновение реагирующих частиц приводит к их взаимодействию. В химическое взаимодействие вступают только активные молекулы, т. е. обладающие энергией, достаточной для осуществления данной реакции. При повышении температуры число активных молекул возрастает, так как нагревание сообщает молекулам необходимую энергию активации, т. е. ту дополнительную энергию, которая приводит к ослаблению химических связей в молекулах реагирующих веществ, а затем и к их разрыву. Зависимость скорости химической реакции от температуры определяется эмпирическим правилом Вант-Гоффа: при повышении температуры на каждые 10о скорость реакции возрастает примерно в 2–-4 раза. Vt где Vt Такое существенное влияние температуры на скорость химической реакции нельзя объяснить только лишь увеличением числа столкновений молекул реагентов. Так как согласно молекулярно-кинетической теории газов средняя скорость молекул пропорциональна квадратному корню из абсолютной температуры, т.е. при повышении температуры, например от 300 до 310 К, средняя скорость молекул возрастет лишь в √310/300 = 1,02 – гораздо меньше, чем требует правило Вант-Гоффа. Объяснить существенное влияние температуры на скорость химической реакции можно используя теорию активации Аррениуса.
Уравнение Аррениуса Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. С. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации. Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию. Опытным путем установлены примерные границы энергий активации для реакций, идущих с соизмеримыми скоростями: если Еа< 50 кДж, то реакция идет с неизмеримо большой скоростью, если же Еа > 100 кДж, то скорость реакции неизмеримо мала. Рассмотрим путь некоторой элементарной реакции А + В ––> С Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения (переходного состояния), называемого активированным комплексом А…В: А ––> А…В ––> B Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е'А выше, чем энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции:
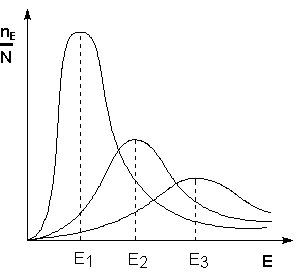
Рис. 1.Энергетическая диаграмма для эндотермической реакции образования продукта АВ из исходных веществ А и В. Если энергия столкновения молекул А и В больше или равна энергии активации Еа, то энергетический барьер преодолевается, и происходит перемещение вдоль координаты реакции r от исходных веществ к продукту. Иначе имеет место упругое столкновение молекул А и В. Вершина энергетического барьера соответствует переходному состоянию (активированному комплексу), в котором связь А–В образовалась частично. Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.):
Рис. 2. Распределение частиц по энергии Зависимость константы скорости реакции от температуры и величины энергии активации описывается – уравнением Аррениуса: k= A e-Ea / RT, где А – предэкспоненциальный множитель, не зависящий от температуры и концентрации и равный числу «удачных» столкновений в единицу времени (т.е. молекулы в момент столкновения должны ориентированы друг относительно друга подходящим образом). Физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения, логарифм константы скорости линейно зависит от обратной температуры: Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
Рис. Зависимость логарифма константы скорости химической реакции от обратной температуры.
Величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
При известных значениях константы скорости реакции при разных температурах:
Уравнение позволяет рассчитать энергию активации процесса по известным константам скорости при разных температурах.
Катализ Катализ - явление изменения скорости химической реакции в присутствии особых веществ – катализаторов. Катализаторы – вещества, которые изменяют скорость химической реакции вследствие многократного участия в промежуточном химическом взаимодействии с реагентами, но после каждого цикла промежуточного взаимодействия восстанавливают свой химический состав. Положительные катализаторы ускоряют реакцию, отрицательные (ингибиторы) замедляют реакцию. Ускоряющее действие катализатора заключается в уменьшении энергии активации катализируемой реакции.
Типы катализа: Гомогенный. Гетерогенный. Автокатализ. Гомогенный катализ – каталитические реакции, в которых реагенты и катализатор находятся в одной фазе. В случае гомогенно-каталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты. Рассмотрим некоторую реакцию А + В ––> С В присутствии катализатора осуществляются две быстро протекающие стадии, в результате которых образуются частицы промежуточного соединения АК и затем (через активированный комплекс АВК#) конечный продукт реакции с регенерацией катализатора: А + К ––> АК АК + В ––> С + К Примером такого процесса может служить реакция разложения ацетальдегида, энергия активации которой EA = 190 кДж/моль: СН3СНО ––> СН4 + СО В присутствии паров йода этот процесс протекает в две стадии: СН3СНО + I2 ––> СН3I + НI + СО СН3I + НI ––> СН4 + I2 Уменьшение энергии активации этой реакции в присутствии катализатора составляет 54 кДж/моль; константа скорости реакции при этом увеличивается приблизительно в 105 раз. Наиболее распространенным типом гомогенного катализа является кислотный катализ, при котором в роли катализатора выступают ионы водорода Н+. Гетерогенный катализ – каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм гетерогенно-каталитических процессов значительно более сложен, чем в случае гомогенного катализа. В каждой гетерогенно-каталитической реакции можно выделить как минимум шесть стадий: 1. Диффузия исходных веществ к поверхности катализатора. 2. Адсорбция исходных веществ на поверхности с образованием некоторого промежуточного соединения: А + В + К ––> АВК 3. Активация адсорбированного состояния (необходимая для этого энергия есть истинная энергия активации процесса): АВК ––> АВК# 4. Распад активированного комплекса с образованием адсорбированных продуктов реакции: АВК# ––> СDК 5. Десорбция продуктов реакции с поверхности катализатора. СDК ––> С + D + К 6. Диффузия продуктов реакции от поверхности катализатора. Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению. Промотирование – увеличение активности катализатора в присутствии веществ, которые сами не являются катализаторами данного процесса (промоторов). Например, для катализируемой металлическим никелем реакции СО + Н2 ––> СН4 + Н2О введение в никелевый катализатор небольшой примеси церия приводит к резкому возрастанию активности катализатора. Отравление – резкое снижение активности катализатора в присутствии некоторых веществ (т. н. каталитических ядов). Например, для реакции синтеза аммиака (катализатор – губчатое железо), присутствие в реакционной смеси соединений кислорода или серы вызывает резкое снижение активности железного катализатора; в то же время способность катализатора адсорбировать исходные вещества снижается очень незначительно. Автокатализ – процесс каталитического ускорения химической реакции одним из её продуктов. В качестве примера можно привести катализируемую ионами водорода реакцию гидролиза сложных эфиров. Образующаяся при гидролизе кислота диссоциирует с образованием протонов, которые ускоряют реакцию гидролиза. Особенность автокаталитической реакции состоит в том, что данная реакция протекает с постоянным возрастанием концентрации катализатора. Поэтому в начальный период реакции скорость её возрастает, а на последующих стадиях в результате убыли концентрации реагентов скорость начинает уменьшаться. Рассмотрим несколько возможных случаев смещения равновесия. В систему aA + bB = mM + nN 1. добавлено исходное вещество. В этом случае
По уравнению изотермы химической реакции получаем: ΔG < 0. В системе возникнет самопроизвольный химический процесс, направленный в сторону расходования исходных веществ и образования продуктов реакции (химическое равновесие смещается вправо). В систему добавлен продукт реакции. В этом случае
Согласно уравнению изотермы химической реакции, ΔG > 0. Химическое равновесие будет смещено влево (в сторону расходования продуктов реакции и образования исходных веществ). 2. Изменено общее давление (для реакций в газовой фазе). Парциальные давления всех компонентов Рi в этом случае изменяются в одинаковой степени; направление смещения равновесия будет определяться суммой стехиометрических коэффициентов Δn. Учитывая, что парциальное давление газа в смеси равно общему давлению, умноженному на мольную долю компонента в смеси (Рi = РХi), изотерму реакции можно переписать в следующем виде (здесь Δn = Σ(ni)прод – Σ(ni)исх):
Примем, что Р2 > Р1. В этом случае, если Δn > 0 (реакция идет с увеличением числа молей газообразных веществ), то ΔG > 0; равновесие смещается влево. Если реакция идет с уменьшением числа молей газообразных веществ (Δn < 0), то ΔG < 0; равновесие смещается вправо. Иначе говоря, увеличение общего давления смещает равновесие в сторону процесса, идущего с уменьшением числа молей газообразных веществ. Уменьшение общего давления газов в смеси (Р2 < Р1) будет смещать равновесие в сторону реакции, идущей с увеличением числа молей газообразных веществ. Необходимо отметить, что изменение концентрации или давления, смещая равновесие, не изменяет величину константы равновесия, которая зависит только от природы реагирующих веществ и температуры.

Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|
 можно рассчитать двумя способами. Записываем уравнение реакции с указанием агрегатного состояния веществ. Подписываем под формулой каждого вещества значения DH
можно рассчитать двумя способами. Записываем уравнение реакции с указанием агрегатного состояния веществ. Подписываем под формулой каждого вещества значения DH  и S
и S  , взятые из справочника:
, взятые из справочника: , кДж/моль 0 0 90,37
, кДж/моль 0 0 90,37 = –RTlnKp.
= –RTlnKp. при Т = 627оС = = 900 К.
при Т = 627оС = = 900 К.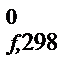 и S
и S  ,кДж/моль –821,22 –1117,71 0
,кДж/моль –821,22 –1117,71 0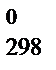 = 2DH
= 2DH 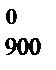 , DS
, DS  = DН
= DН  =
= 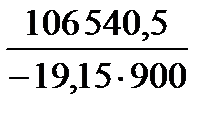 = –6,1816; Kp = 6,583 × 10–7.
= –6,1816; Kp = 6,583 × 10–7. ) = (Kp)2 = 4,334 × 10–13 атм.
) = (Kp)2 = 4,334 × 10–13 атм. = lg1 = 0 и DG
= lg1 = 0 и DG  = –19,15 × Т × lgКp = 0.
= –19,15 × Т × lgКp = 0. = 0:
= 0: =
=  = 1686 К.
= 1686 К.
 = Р1 SnBr
= Р1 SnBr  = 111,55 Дж/К.
= 111,55 Дж/К. и S
и S  , кДж/моль 52,5 90,25 0
, кДж/моль 52,5 90,25 0 и DS
и DS  от температуры
от температуры =
=  = 623,2 К.
= 623,2 К. ) нужно использовать константу равновесия указанного процесса Кр и закон Дальтона.
) нужно использовать константу равновесия указанного процесса Кр и закон Дальтона.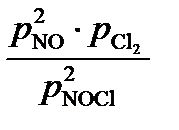 . На основании закона Дальтона Робщ = рNOCl + pNO + pCl
. На основании закона Дальтона Робщ = рNOCl + pNO + pCl  = 1; 1 =
= 1; 1 =  .
.





 × g
× g  ,
,


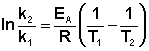

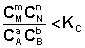 ;
;  ;
;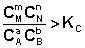 ;
;  ;
;