|
|
Номер механизма соответствует порядковому номеру химической стадии.
В.И.Вигдоровичу и Л.Е.Цыганковой удалось преодолеть математические трудности, хотя это потребовало постулировать ряд не совсем понятных теоретически положений. Однако их подход был проверен многими исследователями нашей страны и получил широкое признание. Рассмотрим используемый подход на конкретном примере. Механизм I. Fе + A FeВадс + Д
Fe(ВД)адс + М Пусть доля поверхности (степень заполнения), занимаемая промежуточным продуктом первой квазиравновесной стадии, равна Q1, второй - Q2. Введем дополнительные допущения: а. ai = bi и, следовательно, равны 0,5. б. Параметр Темкина f для различных частиц один и тот же. в. Адсорбция промежуточных частиц происходят на активных центрах с определенным интервалом значений DGадс, на которых не адсорбируются А, Д, С и М - компоненты раствора. Заметим, что возможность такой «избирательной» адсорбции ОН– и С1– - ионов была показана в ряде работ. Для квазиравновесной электрохимической стадии (50) скорости прямой и обратной реакций равны: i2 = k’’2 Q1 CДехр(aφF/RT) ехр(–bfQ1), i–2 = k’’–2 ехр(–(1–a)φF/RT) ехр[(1–b)fQ2)] В условиях квазиравновесия i2 = i–2, следовательно, можно записать (принимая Q в предэкспоненте постоянной и вводя ее в константы k’’2 и k’’–2): k’2 CД ехр(aφF/RT) ехр(–bfQ1) = k’–2 ехр(–(1–a)EF/RT) ехр[(1–b)fQ2)] Примем отношение Q2/Q1= n, не накладывая на п каких-либо ограничений, кроме оправданного физически неравенства п > 0 . Тогда имеем:
ехр ехр
Для лимитирующей стадии (51) имеем: i3 = k Подставим в последнее уравнение предшествующее, предварительно отметив, что во втором экспоненциальном множителе использован знак «+», а не «-», так как переход адсорбированного комплекса в раствор в процессе этой реакции способствует (стимулирует) дальнейшему протеканию процесса. Здесь имеется определенная натяжка, но ее использование привело к очень интересным экспериментально оправданным результатам. После подстановки имеем: i3 = k Откуда следует i3 = k Из первой квазиравновесной химической стадии следует: Кc = Подставив выражение для Q2 в последнее кинетическое уравнение, получим: i3 = k3 САС И, наконец, окончательно имеем: i3 = k САС Последнее уравнение имеет смысл в области тафелевских наклонов анодной поляризационной кривой от 39 (n/n + 1®1)до 116 мВ (порядок реакции ионизации по компоненту Д®0), то есть в общем случае можно записать: 39 где Принципиальным отличием полученного кинетического уравнения от всех приводимых ранее является: 1. Возможность изменения величины bа в широких пределах без смены механизма процесса. 2. Возможность изменения порядка реакции по реагирующим частицам в широких пределах также без смены механизма процесса. 3. Возможность объяснения дробных величин nд. Наличие функциональной зависимости между nд и bа, что является новым критерием механизма процесса (рис. 16).
Рис. 16. Графическое изображение связи nд с bа.
Найдем связь между порядком реакции по веществу (частице) Д и bа nД = bа =2,3×2 bа =2,3×2 Окончательно имеем: bа =2,3×2 nД = Рассмотрим несколько иной, но вполне возможный механизм ионизации железа. Схема его внешне весьма незначительно отличается от предложенного выше и определяемого стадийностью (49) -(51).
Механизм II. Fe + A FeВ+адс + Д
Fe(ВД)+адс + М
Их различие заключается в том, что в последнем первая стадия является электрохимической, а вторая - химической. Это казалось бы небольшое изменение ведет к существенным различиям в суммарном кинетическом уравнении процесса, который имеет вид (предлагаем читателям получить его самостоятельно): i3 = k CДCМ C Найдем связь между порядком реакции по компоненту А и та-фелевским наклоном. nA = 0,5n, n = 2nA (где nA – порядок реакции по компоненту А). bа =
2nA bа + bа = nA = Уравнения, связывающие порядок реакции по компоненту А и тафелевский наклон, идентичны с соответствующими зависимостями для механизма I (стадии (49) - (51)). Найдем связь между порядком реакции по компоненту С и bа. nС = –0,5n, n = 2nC bа =
Так как в области активного растворения железа тафелевский наклон больше 0, то всегда пс < bа – 2nC bа = Так как bа всегда больше 0 (активное растворение металла), то пс < Представляет интерес выяснить соответствующие связи между nQH–и bа для механизма Хойслера. Полученное нами с использованием изотермы Темкина кинетическое уравнение имеет вид: ia = k C С учетом связи п и nOH– имеем: bа = В этом случае легко объяснить результаты стационарных и нестационарных измерений, не прибегая к необходимости изменения механизма процесса (см. механизм К. Хойслера). И, наконец, предлагаем проанализировать механизма III с использованием изотермы Темкина самостоятельно.Он отличается от предшествующих двух тем, что в нем первая и вторая стадии являются электрохимическими одноэлектронными, а лимитирующая последняя - химической.
АНОДНОЕ ПОВЕДЕНИЕ ЖЕЛЕЗА В РАСТВОРАХ С ВЫСОКОЙ ИОННОЙ СИЛОЙ Все рассмотренные ранее механизмы (Фрумкина, Бокриса, Хойслера и др.) предполагают, что ионизация железа протекает с участием гидроксид-ионов, a (d1gia/d1gCH+) < 0. Это показано для сульфатных (рН – 1,2 - 4,9), хлоридных (рН – 1,1 - 3,1) и фосфатных (рН – 1,2 - 4,0) растворов. Однако дальнейшее увеличение концентрации протонов меняет характер зависимости. В сильнокислых средах, содержащих поверхностно-активный анион, порядок реакции ионизации железа по ионам водорода в определенном интервале СH+ становится равным нулю (рис. 17).
Рис. 17. Зависимость скорости анодного растворения железа в солянокислых водных растворах (при постоянной ионной силе) от активности ионов водорода (по данным американских и немецких исследователей).
Далее с ростом СH+ величина (d1gia/d1gCH+)Ci, E приближается к 1 или 2, что наблюдается в растворах с высокой и фиксированной ионной силой (J = 5 - 6). Концентрационный интервал с nH+= 0 наблюдается не всегда. Его наличие определяется природой и составом электролита. Дарвиш, Хилберт, Лоренц и Россваг объяснили наличие nH+ > 0 переходом к новому механизму растворения, который можно выразить следующей системой реакций: Fe + С1– « (FeС1)адс +е; (FeС1)адс + Н+ « (FeClH)+адс (ионные пары); (FeClH)+адс ® FeCl+адс + Н+ +е.
Мкафферти и Хакерманн для объяснения кинетических параметров в концентрационном интервале с nH+ > 0 предлагают механизм: Fe + Н2О « Fe(Н2О)адс; Fe(Н2О)адс + С1– « FeС1–адс+ Н2О; FeС1–адс+ Н+ «FeС1–Н+ (ионные пары); FeС1–Н+ ® FeCl+ + Н+ +2е. Как видно, все исследователи для объяснения кинетики процесса с nH+ > 0 прибегают к представлениям о возникновении ионных пар на поверхности металла. С использованием приведенных и других опубликованных экспериментальных данных следует полагать наличие двух критических концентраций протонов ( К. L. М. Если это явление носит достаточно общий характер, то абсолютная величина предельных концентраций протона должна определяться природой растворителя и электролита и ионной силой раствора. Тогда в ряде случаев не исключено равенство В этанольных растворах (10 % Н2О) с ионной силой 7 (рис. 18) наблюдаются все три концентрационные интервала. Величины концентраций излома Замена этанола на этиленгликоль существенно не изменяет качественного характера зависимости. Кроме того, получено, что величина (d1gia/d1gCС1–)Ci, j для железа в этанольных растворах колеблется в пределах 0,4 - 0,85, в этиленгликолевых средах близка к 1 независимо от содержания воды. Ионизация железа в этанольных растворах НС1 протекает по механизму 1, который для nH+ < 0 может быть детализирован посредством схемы: Fe + « Fe(RО–)адс + H+ (55) Fe(RО–)адс + C1– « (FeRОC1–)адс + e (56) (FeRОC1–)адс ® FeCl+ + RO– + e (57)
Рис. 18. Зависимость скорости анодного растворения железа при постоянном потенциале электрода от концентрации ионов водорода (солянокислые растворы, постоянная ионная сила, Н2 - атмосфера, комнатная температура),
В общем виде кинетическое уравнение механизма 1 имеет вид: iа = k CА C Применительно к схеме (55) - (57) оно упрощается: iа = k СROH C Далее примем, что при достижении Fe + ROH « Fe(ROH)адс; (58) Fe(ROH)адс + С1– « (FeROHCl)адс + е; (59) (FeROHCl) адс ® FeCl+ + ROH + е . (60)
Тогда на основании общего кинетического уравнения имеем: iа = k С С достижением критической концентрации Рассмотрим несколько подробнее ионные ассоциации в растворе. При растворении вещества с ковалентной связью за счет сольватации возможна определенная поляризация его молекул, при которой происходит переход от полярной ковалентной связи к ионной. Говорят, что происходит ионизация вещества, а растворители, в которых наблюдается этот процесс, называются ионизирующими. Если процесс на этом останавливается, то в растворе в значительном количестве будут присутствовать ионные пары. При значительном ослаблении электростатического взаимодействия происходит электролитическая диссоциация ионных пар на отдельные ионы. Подобные растворители называют диссоциирующими. Кроме ионных пар, которые существуют по современным воззрениям в концентрированных растворах и образованию которых способствует понижение диэлектрической проницаемости растворителя, возможно образование ионных тройников типа К+А–К+ и А–К+ А– и более сложных образований (К+ - катион, А– - анион). Большинство исследователей считает, что ионные пары не участвуют в переносе тока. Впервые концепция ионных пар была предложена Бьеррумом (Дания) в 1926 г. Причем, в настоящее время исследователи считают, что встречаются ионные пары трех типов (В.И.Словецкий. Ионные пары в органической химии. М: Изд-во "Знание", 1976. № 8): 1. Контактные, когда между ионами пары нет молекул растворителя. 2. Растворитель - разделенные (или рыхлые). При образовании пары оба иона сохраняют индивидуальные первичные сольватные оболочки, вторичная у них общая. 3. Ионные пары с частичным участием растворителя. При образовании ионной пары между ионами остается одна молекула растворителя, через которую они связаны. Изменение свободной энергии взаимодействия двух ионов в рассматриваемой электростатической модели выражается зависимостью: DG1,2 = z1×z2×e2/er1,2 где r1,2 – расстояние между ионами, e - диэлектрическая проницаемость среды, zi - заряд иона. DG1,2 = NA z1×z2×e2/er1,2= 1385 z1×z2/er1,2кДж/моль, NA - число Авогадро. Подтверждением применимости такого подхода является соблюдение линейной зависимости между логарифмами констант диссоциации ионных пар и 1/e. Это же указывает на независимость В теории растворов наибольшее признание получила теория Дебая - Гюккеля, основные положения которой следующие: 1. В водных растворах сильные электролиты полностью диссоциированы. 2. Коэффициенты активности отдельных (неассоциированных) ионов зависят от общей концентрации ионов в растворе, выраженной через ионную силу (J = 3. Подвижность свободных ионов также зависит от их общих концентраций в растворе в результате влияния ионной атмосферы. 4. Первичный солевой эффект обусловлен зависимостью коэффициентов активности свободных ионов от общей концентрации ионов в растворе. В.А.Пальм в монографии «Основы количественной теории органических реакций» (Л.: Изд-во "Химия", 1977) решительно отказывается от теории Дебая-Гюккеля, указывая, что «наиболее слабым ее местом следует считать выделение водных растворов электролитов из числа всех остальных». Например, тезис о полной диссоциации относится именно к водным растворам. Само это положение возникло, скорее всего, как результат невозможности описания концентрационных зависимостей электропроводности водных растворов сильных электролитов, исходя из простого закона разбавления Оствальда. Или, говоря более общим языком, невозможности однозначного описания их диссоциации на основе закона действующих масс даже после введения коэффициентов активности. По мнению Пальма, действительный источник затруднений - тот факт, что ионные пары (или другие электронейтральные ионные агрегаты) не могут участвовать в электропроводности. В его работе (Пальм В.А., Карелсон М.М. // Реакционная способность органических соединений, 1974. Т. 11. № 2 (40). С. 519-533) показано, что в диапазоне от 0,2 – 0,5 М до насыщения электролитов все ионы входят в состав ионных агрегатов. Доказательство основано на количественной интерпретации положительных отклонений от линейности в разбавленных растворах электролитов в координатах Таким образом, независимо от той или иной концепции, возможность возникновения ионных ассоциациатов является доказанным фактом, следовательно, образование в высококонцентрированных кислых спиртовых растворах частиц Н+С1– и Н+С1–Н+ вполне допустимо. Оно тем более вероятно в плотной части двойного электрического слоя на твердых электродах, где существенно понижена e. Внедрение частиц типа Н+А– в плотный слой, по мнению Я.В.Дурдина и Е.Г. Цвентарного, можно представить как результат перехода Н+С1–из раствора, так и за счет присоединения иона водорода к уже адсорбированному аниону. Если встать на подобную точку зрения, то не трудно в рамках единого механизма объяснить наличие интервала, в котором nH+ > 0 . Для этого необходимо принять, что процесс протекает по схеме: Fе + Н2О « Fе(Н2О)адс; (61) Fe(H2O) адс +Н+С1– « (FeH2OClH+) адс +e; (62) (FeH2OClH+) адс ® FeCl+ +H3O+ + e. (63) В соответствии со схемой (61) – (63) для фиксированного состава растворителя кинетическое уравнение принимает вид iа = k C Если в ионном ассоциате 2 иона водорода, то имеем iа = k C Если концентрационный интервал, в котором подавляется диссоциация адсорбированной воды, совпадает с областью существования ионных двойников, то
АНОДНОЕ РАСТВОРЕНИЕ МЕТАЛЛОВ ПРИ
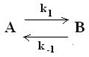
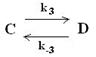
Рассмотрим кинетику стадийного процесса анодной ионизации металла, когда скорости отдельных стадий соизмеримы и, следовательно, предшествующие стадии нельзя считать квазиравновесными. Однако предварительно выведем кинетическое уравнение для стадийного обобщенного химического процесса, протекающего по схеме:
(64);
(65);
(66);
Из принятого условия следует, что все последовательные стадии обратимы. При стационарных условиях скорости процессов (64), (65) и (66) одинаковы, то есть V1 = V2 = V3. Пусть а - активность соответствующего компонента. V1 = k1 aA – k–1 аB. (67) V2 = k2 aB – k–2 аC. (68) V3 = k3 aC – k–3 аD. (69) Из уравнения (67) выразим аB: аB = Из уравнения (68): aC = Подставив в последнее выражение значение аB, получим: aC = aC = V3 = V = V k–2 k–1 + k3 k2 V + k3 k–1 V = k3 k2 k1 aA – k–3 k–2 k–1 aD; V = V =
Так как суммарная реакция со значительной скоростью протекает слева направо, то ki >> k–i. Тогда V = Полученную зависимость (70) можно применять для вывода кинетического уравнения стадийного электрохимического процесса, например, протекающего по схеме, соответствующей механизму 1. Fе + A FeВадс + D
Fe(ВD)+адс + М В дальнейших выводах будем считать, что степень заполнения поверхности промежуточными продуктами невелика и DGадс ¹ j (Q). aFe = 1, ai = bi = 0,5. Активности частиц A, С, D, M и BDC достаточно велики и их изменением можно пренебречь, тогда V1 = k1 aA; V–1 = k–1 аFeВадс аС; V2 = k2 ехр V–2 = k–2 ехр V3 = k3 ехр V–3 = k–3 ехр Выразим суммарную скорость процесса в электрических единицах. При этом лишь несколько изменятся абсолютные значения и размерности констант. Подставим величины констант скоростей в уравнение (70). Причем константу объединим с экспоненциальным членом, так как последняя величина не зависит от концентрации реагирующих частиц. Предварительно отметим, что если при выводе уравнения (70) учесть концентрацию компонентов реакции, участвующих в последующих стадиях, то их концентрации войдут в формулу в виде произведения с соответствующей константой скорости: iа = iа = Пусть первая стадия протекает значительно медленнее, чем две последующие: k1 << k2, k1<< k3, k–1<<k–3, k–1<k1, k–1<<k2. Тогда все члены с k–1 в знаменателе очень малы по сравнению с другими и ими можно пренебречь Окончательно получаем: iа = iа = Как и следовало ожидать, в этом случае скорость реакции не зависит от потенциала. Если лимитирующей является вторая стадия, то k2 и k–2 очень малы и членами в знаменателе с этими константами скоростей можно пренебречь. Тогда имеем: iа = iа = Наконец, если лимитирует третья стадия, то в знаменателе общего кинетического уравнения можно пренебречь членами с k3 и k–3: iа = iа = Так как принято α = b = 0,5, то: iа = Рассмотрим двустадайный процесс со сравнимыми скоростями последовательных стадий: A В V = k1 aA– k–1 аВ; аВ = V = k2 aВ– k–2 аС; V = V
V = V =
Выведем кинетическое уравнение для процесса, протекающего по схеме: Fе + A FeВ+адс + D стадии которого характеризуются сравнимыми скоростями, а условия адсорбции соответствуют изотерме Темкина:
В этом случае в полулогарифмических координатах на поляризационных кривых отсутствуют линейные участки.
ПАССИВНОСТЬ МЕТАЛЛОВ
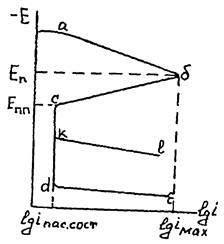
Активное растворение металлов, которое мы рассматривали до сих пор, предполагает рост скорости ионизации по мере сдвига потенциала электрода в положительную сторону. При этом часто наблюдается линейная зависимость, представленная в полулогарифмических координатах. Однако в определенной области потенциалов происходит резкое торможение анодной реакции по мере роста потенциала, а затем и независимость i от φ. Это удобно рассмотреть на примере полной анодной поляризационной кривой (рис. 19). аб - участок активного растворения металла; бс - участок, на котором металл переходит в пассивное состояние, cd - участок пассивности, Еn - потенциал пассивации, при достижении которого начинается пассивация металла, Епп - потенциал полной пассивации (Фладе - потенциал, при котором металл переходит в пассивное состояние); dе - участок перепассивации, на котором, как правило, начинается новый анодный процесс (транспассивное состояние), связанный с переходом металла в раствор в высшей степени окисления. В присутствии некоторых анионов, прежде всего, ионов хлора, возможно нарушение пассивного состояния до наступления перепассивации (участок kl) Это явление, как правило, приводит к образованию питтингов, очевидно, за счет локализации анодной реакции на небольшой площади. Облегчение наступления пассивного состояния за счет изменения внешних условий (рН, анионный состав, ионная сила раствора) выражается в сдвиге потенциала электрода в отрицательную сторону и снижении тока пассивации. Ток в пассивном состоянии может при этом не изменяться, увеличиваться или уменьшаться (рис. 20).
Рис.19. Общий вид анодного участка поляризационной кривой металла. Пояснения в тексте.
Рис. 20.Некоторые возможные изменения на поляризационной кривой в различных условиях ионизации металла.
Важную роль играет область потенциалов (СД), в которой металл пребывает в пассивном состоянии. Чем она протяженнее по потенциалу, тем устойчивее пассивное состояние. В ряде случаев может наблюдаться несколько максимумов пассивации, например, на никеле в растворе с составом электролита 1 м/л НС1О4+0,5 м/л H2SO4 (рис 21) или железе в боратном растворе Н3ВО3 (рН = 7,4) с добавкой 0,1 моль-экв/л Na2SО4.
Рис. 21. Анодная поляризационная кривая никеля (а) в электролите 1 моль/л НС1О4+ 0,5 моль/л H2SO4 и железа (б) при рН = 7,4 (боратный электролит с 0,05 моль/л сульфата натрия).
Механизм пассивации металлов, видимо, очень сложен. В настоящее время существует две теории пассивности: пленочная и адсорбционная. Прежде всего (по В.М. Новаковскому), они различно решают вопрос о том, на какой процесс при растворении пассивного металла расходуется анодный ток и каким путем катионы металла попадают в раствор. Исходя из адсорбционной теории, несколько упрощенно излагая ситуацию, укажем, что анодная реакция на пассивном металле, как и на активном, связана с переходом в раствор катионов из металлической решетки. Пассивация же сводится к повышению перенапряжения этого перехода на металле, поверхность которого адсорбирует кислород или молекулы воды кислородным концом. По пленочной теории малая скорость растворения пассивного металла объясняется тем, что при пассивации совершенно меняется природа процесса. Вместо образования гидратированных катионов ток, по этим представлениям, расходуется на анодное образование пассивирующего оксида (М.Фарадей тоже так раньше других интерпретировал пассивность железа, впервые же пассивность железа в азотной кислоте наблюдал М.В.Ломоносов), а катионы попадают в раствор только за счет последующего растворения этого оксида, которое рассматривается как обычная химическая реакция. Пассивные участки кривой нельзя зафиксировать посредством гальваностатической методики снятия поляризационной кривой, так как одной и той же величине тока на последней соответствует несколько значений потенциала (рис. 21а). Поэтому используется по-тенциостатическая методика, по которой задают фиксированные значения потенциала и измеряют соответствующие им величины тока. Так как токи обычно длительное время изменяются, то такие кривые не бывают вполне стационарными. Обычно величина тока фиксируется через определенный промежуток времени (50 - 60 минут) после наложения потенциала. На практике часто весь интервал потенциалов проходят за определенное время с постоянной скоростью развертки потенциала, получая потенциодинамические кривые. Ряд исследователей, как уже отмечалось, в отсутствие солевой пассивности (типа Рb в H2SO4) объясняют пассивное состояние образованием фазовой пленки, другие - посредством адсорбционной пленки за счет взаимодействия поверхностных атомов металла с молекулами Н2О или ионами ОН–. Фазовый оксид: 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|
 FeВадс + C; (49)
FeВадс + C; (49) Fe(ВД)адс + е; (50)
Fe(ВД)адс + е; (50) Fe2+ + ВДС + e . (51)
Fe2+ + ВДС + e . (51) = ехр(–φF/RT) ехр(fQ2–bfQ2+ bf
= ехр(–φF/RT) ехр(fQ2–bfQ2+ bf  );
); =
=  ехр(φF/RT)
ехр(φF/RT) =
=  = ln [
= ln [ 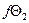 =
=  ln[
ln[  Q2 Cм ехр(aφF/RT) ехр(bfQ2).
Q2 Cм ехр(aφF/RT) ехр(bfQ2).

 ; Q2 =
; Q2 =  ;
; Cм С
Cм С  ехр
ехр 

 2,3
2,3 
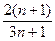 - кажущийся коэффициент переноса. Вне указанных пределов bа п < 0, что лишено физического смысла.
- кажущийся коэффициент переноса. Вне указанных пределов bа п < 0, что лишено физического смысла.
 ; n =
; n =  .
.

 - гиперболическая зависимость.
- гиперболическая зависимость. – тоже, естественно, гипербола (рис. 16).
– тоже, естественно, гипербола (рис. 16). FeB
FeB  + C + е (52)
+ C + е (52) Fe(ВД)+адс ; (53)
Fe(ВД)+адс ; (53) C
C  ехр
ехр 
 ; bа =
; bа =  ; bа =
; bа = 
 ;
; –
–  ; nA =
; nA =  b
b  –
–  ;
; ; bа =
; bа =  ;
; ехр
ехр 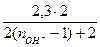
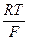 ; bа =
; bа =  .
.
 и
и  ), относительно которых по величине nH+ можно выделить три концентрационных интервала:
), относительно которых по величине nH+ можно выделить три концентрационных интервала: <
< 
 C
C  ехр
ехр  ).
). C
C  .
. C
C  .
. =
= 
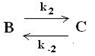
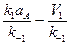 ,
,


 - k-3aD
- k-3aD ;
;
 =
= 
 <<
<<  и во втором сомножителе знаменатель близок к 1. Тогда
и во втором сомножителе знаменатель близок к 1. Тогда (70)
(70) FeВадс + C;
FeВадс + C; Fe(В D)+адс + е;
Fe(В D)+адс + е; аFeВадс аD;
аFeВадс аD;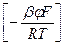 а(FeВD)+адс;
а(FeВD)+адс;

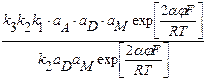 ;
;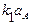 ;
;


 .
. .
. В;
В; ;
; ;
; =
= 
 =
= 
 ;
; ;
; ;
;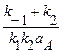 ; V =
; V =  .
. ;
; ;
; ;
;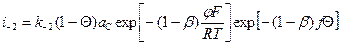 ;
; ;
; ;
;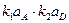 = M;
= M; = N;
= N; = L.
= L.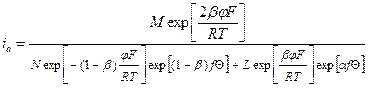 ;
;
 ;
;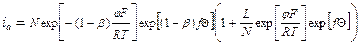 .
.