
|
|
Механизм Я.М.Колотыркина и Г.М. ФлориановичОн интерпретирован в условиях анодной ионизации железа в кислых сульфатных средах и включает как последовательные химические и электрохимические, так и параллельные стадии. Fe + Н2О « (FеOH–)адс + H+; (42) (FеOH–)адс « (FеOH)адс +e; (43) (FеOH)адс + HSO4– ® FeSO4 + Н2О + е; (44а) (FеOH)адс + SO42– ® FeSO4 + ОН– + е.. (44б)
С учетом параллельно протекающих лимитирующих стадий (44а) и (44б) можно записать: iа = k’
Учитывая квазиравновесие стадии (43), получим: φ = φ Откуда после несложных неоднократно проводимых ранее преобразовании имеем
Сочетание кинетического уравнения лимитирующей стадии и выражения для iа = k44 k43 Состояние квазиравновесия реакции (42) позволяет записать: Kа = В водном растворе Сочетая последнее выражение с кинетическим уравнением, полученным с учетом стадий (43) и (44), окончательно имеем: iа = k Таким образом, кинетическое уравнение, полученное с учетом ионизации металла по механизму Колотыркина - Флорианович, удовлетворительно коррелирует с экспериментальными данными: bа =35 мВ, пA– = 1, пH+ = – 1 (область рН - 1...5). В качестве еще одного примера рассмотрим результаты исследования кинетики анодного растворения меди в кислых хлоридных метанольных растворах и предложенный авторами механизм процесса (В.И.Вигдорович, Л.Е.Цыганкова, 1976 г.). Работа проведена методом снятия стационарных анодных поляризационных кривых. Порядок реакции по ионам водорода получен в растворах с составом электролита: XmHC1+ (J-X)m LiC1 где J - ионная сила раствора, равная 1,0; 2,5 и 4,0. Порядок реакции по ионам хлора получен в хлоридно-перхлоратных растворах с составом электролита 0,1mHC1+ Xm LiC1+ (3,9 – х) LiC1О4, ионная сила равна 4.
Рис. 13. а) Тафелевские участки анодных поляризационных кривых процесса ионизации меди в кислых хлоридных метанольных растворах с постоянной ионной силой и СС1– = const. Цифры на кривых – СН+, моль/л; б) схематическое изображение зависимости скорости анодной ионизации меди от концентрации ионов Н+ или С1– при потенциале Еа и постоянной ионной силе в метанольных хлоридных или хлоридно-перхлоратных растворах.
Из экспериментально полученных зависимостей, изображенных на рис. 13а, строили зависимость типа показанной на рис. 13б. Тангенс угла наклона последней прямой, как указывалось выше, представляет собой порядок реакции ионизации меди по соответствующим частицам (ионам). Полученные в работе данные обобщены в таблице (1). Таблица 1. Порядок анодной реакции ионизации меди по ионам водорода, nН+, и хлора, nС1–, и величины bа, мВ тафелевского участка анодных поляризационных кривых в растворах CH3OH
* СНС1 - 0,10 - 0,99 моль/л.
Все исследования проведены с использованием вращающегося дискового электрода в отсутствие диффузионных ограничений. Независимыми опытами было показано, что эффективный заряд переходящих в раствор ионов меди равен 1, величина порядка реакции ионизации по ионам Сu2+ равна 0. Полученные кинетические параметры удовлетворительно интерпретируются посредством следующего механизма ионизации меди: Сu + С1– « (СuС1)адс +е; (45а) Сu + Н+С1– « (CuHCl)+адс +е; (45б) Сu + Н+С1–Н+ « (CuH2Cl)2+адс +е. (45в) Или обобщенно Сu + (НnС1l)Z « (СuНnС1l) где z и z + 1 - заряды соответственно ионного образования и промежуточного адсорбционного комплекса, п меняется от 0 до 2. Далее протекает лимитирующая химическая стадия: (СuНnС1l) Для стадии (46) запишем кинетическое уравнение: i46 = k46 Из предшествующей квазиравновесной стадии следует:
Сочетая кинетическое уравнение лимитирующей стадии и выражение для iа = k или iа = k которое допускает экспериментальную проверку. При малой концентрации НС1 ионные двойники и тройники не играют существенной роли (nн+ =0) и квазиравновесная стадия протекает по уравнению (45а). nн+ = 2 говорит о том, что реализуется преимущественно процесс (45 в). Обобщая сказанное, можно записать iа = (k45a +k45б Исходя из величины пС1–,р = 1 или 2. Введение воды способствует разрушению ионных двойников и тройников, особенно в растворах с малой ионной силой. Поэтому в присутствии 10 мас.% Н2О 0 < nн+ < 1. Когда k45a >> (k45б больше k45в Из приведенных результатов следует, что в хлоридных метанольных растворах ионы однозарядной меди существуют в виде комплексных ионов СuС12– и СuС132–, что имеет место и в воде. Все рассмотренные механизмы ионизации металлов базируются на представлении об образовании в ходе процесса промежуточного адсорбционного комплекса (ПАК). Между тем структура ПАК изучена мало. Согласно одним данным, ПАК, например, на железе представляют собой комплексы с переносом заряда (ПКПЗ) типа: Ме-Н2О-Аn- или ацидокомплексы Ме-Аn-, где Аn- - анионы кислот (SO42–, HSO4–, C1–, I–). Доля перенесенного заряда составляет 0,05 - 0,13 заряда электрона. Вода в ПКПЗ является донором электронов, претерпевающих в основном состоянии частичный, а в возбужденном - полный перенос в свободную зону проводимости металла (выше уровня Ферми). Помимо данных спектроскопии, предполагаемый количественный состав ПАК на железе подтверждается порядками реакции ионизации по анионам и равной единице величиной dlgia/dpH. Первый порядок по растворителю при анодном растворении железа наблюдался в кислых хлоридных растворах смешанных растворителей в системах СН3ОН - Н2О и С2Н5ОН - Н2О. Используя понятия химии комплексных соединений (КС), такие ПАК формально можно представить в виде Ме[H2O]An– (А) Здесь активный центр поверхности металла, обозначенный как Me, играет роль комплексообразователя, молекула Н2О - лиганда внутренней, а An– - внешней сферы КС. В случае адцидокомплексов трудно представить, что в безводном варианте ПАК анион лишен гидратной оболочки. Более вероятно замещение лигандов внешнесферными ионами с образованием ПАК вида: Ме[An–]H2O, что, по существу, является изомерным превращением КС. Частицы внутренней сферы поверхностного комплексного образования определяют связь ПАК с поверхностью металла, частицы, образующие внешнюю сферу, - с объемом раствора. Если энергетически более прочны первые связи, - поверхностные комплексные образования выступают в качестве ингибиторов ионизации металла; если же энергия связи частиц внешней сферы с объемом раствора более высока, чем внутренней с поверхностью металла, ПАК выступают в качестве активатора процесса ионизации. Отмеченную смену характера действия частиц, участвующих в процессе ионизации, можно представить в виде равновесия: ПАК - активатор « ПАК - ингибитор. Последнее равновесие, в зависимости от условий протекания процесса, может смещаться в ту или иную сторону. Некоторое недоумение может вызвать тот факт, что координационное число (к.ч.) комплексообразователя часто равно 1, чего не наблюдается в химии координационных соединений. Однако, во-первых, это вполне можно объяснить особенностями, вносимыми существованием КС на границе раздела фаз. Во-вторых, можно принять, что активный центр представлен несколькими атомами металла, достаточно прочно связанными с кристаллической решеткой, и КС следует рассматривать как определенное подобие многоядерного комплекса, в котором каждый входящий в его состав атом адсорбирует молекулу растворителя в качестве лиганда внутренней сферы. Тогда координационное число такого КС, играющего роль ПАК, может оказаться существенно отличным от единицы, характеризующей формальный экспериментально наблюдаемый порядок процесса по молекулам растворителя и предстающей собой отношение числа молекул сольвента в ПАК к числу атомов металла в составе активного центра. Следовательно, вместо поверхностного адсорбционного комплекса типа (А) для внутренней сферы имеем: [Меn(Сольвент)m] (В) с т > 1 и целочисленным. Наличие на поверхности железа биядерного комплекса было принято, как ранее отмечалось, Хойслером. Такой же подход использован при интерпретации анодного растворения свинца в растворах КОН, где авторы исследования принимают ПАК в виде Рb2(ОН)2. Очевидно, что в последнем случае в качестве лигандов ПАК выступают гидроксид-ионы, внешняя же сфера состоит из молекул воды, играющих роль гидратной оболочки. Казалось бы, комплексные соединения типа (В) можно рассматривать как внутрисферные. Однако это не так. В них имеется и внешняя сфера, частицами которой, как уже отмечалось, также являются молекулы растворителя. Однако последние менее прочно связаны с активным центром, чем лиганды, и одновременно с большей энергией взаимодействуют с растворителем в объеме жидкой фазы. Следует отметить ряд интересных экспериментальных фактов, наблюдаемых при анодной ионизации ряда металлов, некоторые из которых уже приводились ранее. Порядок реакции анодной ионизации меди в хлоридных растворах по ионам хлора обычно равен 2 или 3, то есть в этом случае ПАК имеет вид [CuCIn]n–(H2O)k с n = 2 или 3, что удовлетворительно коррелирует со структурой стабильных комплексных ионов CuCl2– или СиС132–, существующих в объеме раствора. Так что аналогия между структурой КС, играющих роль ПАК, и структурой КС в объеме раствора имеет право на существование. Подтверждением этого являются также результаты ряда других исследований. В частности, порядок анодного растворения цинка по хлорид-ионам в хлоридных водно-диметилацетамидных растворах равен 6, что указывает на образование ПАК типа [ZnCl6]6–(H2O)k и удовлетворительно коррелирует с образованием стабильных комплексов цинка подобного состава, существующих в объеме раствора. С переходом от диметилацетамида к диметилформамиду наблюдается изменение координационного числа ПАК по хлорид-ионам [ZnCl4]4–(H2O)k, но величина к.ч. вновь соответствует хорошо известной для стабильных комплексов ионов цинка Несомненно, состав ПАК является функцией многочисленных факторов: качественного и количественного состава раствора, потенциала электрода. В определенных условиях нельзя исключить влияние гидродинамических условий в приэлектродном слое. Принципиальное значение имеет учет энергетической неоднородности поверхности металла, к рассмотрению которой мы и переходим.
УЧЕТ ЭНЕРГЕТИЧЕСКОЙ НЕОДНОРОДНОСТИ ПОВЕРХНОСТИ МЕТАЛЛА Использование для описания кинетики предшествующих электрохимических стадий изотермы Генри, как уже отмечалось, соответствует постулату, что доля поверхности, занятая промежуточными адсорбированными частицами, невелика, во всяком случае, существенно менее 20 %, а задействованные в процессе активные центры энергетически равноценны. Это приводит к ряду сложностей в интерпретации экспериментальных данных: - тафелевские наклоны линейных участков анодных поляризационных кривых не могут принимать значения в интервале 116 > bа > 58 мВ. Экспериментально же наблюдаются величины 70 - 100 мВ, что трудно объяснить с этих позиций. Приходится постулировать значительное изменение величины коэффициента переноса анодного процесса, который в ряде случаев вообще должен принимать физически необоснованные значения; - порядки анодной реакции по реагирующим частицам могут иметь только целочисленные значения. Экспериментально очень часто (особенно в концентрированных растворах сильных кислот) наблюдаются дробные величины. Для интерпретации подобных явлений можно использовать постулат Темкина, согласно которому поверхность ряда металлов (прежде всего железа) является равномерно-неоднородной. Такая картина может иметь место, если степень заполнения поверхности металла промежуточными адсорбированными частицами находится в пределах Q = 20.....80%. Адсорбция на такой поверхности описывается посредством логарифмической изотермы или изотермы Темкина: Q = где а0 - константа адсорбционного равновесия, Р - давление адсорбата. Используя вместо давления адсорбата его равновесную концентрацию, можно записать: Q = Если значение параметра f достаточно велико, то должна существовать промежуточная область заполнений, для которой а0С >>1 и а0С ехр(-f) <<1. Тогда уравнение изотермы упрощается: Q = f - параметр Темкина, характеризующий свободную энергию адсорбции. Для этого случая DG DG Причинами, приводящими к изменению свободной энергии адсорбции с заполнением, являются следующие: - собственная (биографическая) неоднородность поверхности, которая однако не позволяет полностью объяснить наблюдаемые эффекты, хотя концепция активных центров широко используется в катализе;
Рис. 14. Схематическое изображение ступенчатого характера зависимости DGадс от степени заполнения поверхности электрода частицами адсорбата в условиях реализации изотермы Темкина.
- эффект поверхностного взаимодействия. Гориуги и его сотрудники рассмотрели вопрос об изменении теплоты адсорбции под влиянием сил отталкивания между молекулами адсорбата. Они показали, что теплота адсорбции линейно связана с заполнением. Однако этот эффект недостаточно велик и только им наличие подобной зависимости объяснить нельзя; - эффект индуцированной неоднородности, сформулированный впервые Бударом. Понижение теплоты адсорбции с ростом заполнения объясняется влиянием адсорбции на работу выхода электрона из металла. Последняя модель является наиболее приемлемой и в наибольшей мере обусловливает линейность зависимости DG Известные электрохимики Конуэй и Гилеади в одной из своих публикаций пишут: «Вызывает удивление тот факт, что интерпретация большого числа экспериментальных данных по кинетике электродных процессов проводится до сих пор исключительно на основе изотермы Ленгмюра, несмотря на то, что теоретические подходы и соответствующие модифицированные уравнения скорости с использованием изотермы Темкина предложены несколько десятков лет назад А.Н Фрумкиным». (А.Н Фрумкин - выдающийся советский ученый, академик, своими работами способствовавший рождению электрохимической кинетики в ее современном виде. Еще при жизни ученого ведущие электрохимики США называли его электрохимиком мира № 1.) Изотерма Ленгмюра описывает любые значительные степени заполнения поверхности металла, но справедлива лишь для энергетически однородной поверхности в условиях мономолекулярной адсорбции. Однако вернемся к изотерме Темкина. Для простой стадии типа: Ме + Х– кинетические уравнения для прямой и обратной реакций имеют вид: i1 = k1’(1–Q) Cх- ехр(bφF/RT) ехр(–afQ); i–1 = k1’Q ехр(–(1–b)φF/RT) ехр[(1–a)fQ)], где СМеХ = kQ . Так как концентрация промежуточных частиц влияет на скорость этой реакции прежде всего через экспоненциальные члены, зависящие от Q, то изменением Q в предэкспоненте можно пренебречь, а уравнения упростить:
i1 = k1 Cх- ехр(bφF/RT) ехр(–afQ), i–1 = k–1 ехр(–(1–b)φF/RT) ехр[(1–a)fQ)]
Члены ехр(–afQ) и ехр[(1–a)fQ)] учитывают влияние на скорость процесса энергетической неоднородности поверхности и изменения DG0 с ростом Q (рис. 15).
Рис 15. Схема, поясняющая характер зависимости DG
Рассмотрим использование концепции энергетической неоднородности поверхности на базе изотермы Темкина на конкретном примере, опубликованном в одном из электрохимических журналов. Авторы рассматривали механизм процесса, описываемый схемой: Me + А МеВадс + Д Стадия (47) является квазиравновесной, а (48) - лимитирующей. Для квазиравновесной стадии можно записать: i1 = k1’ (1–Q) ехр(–afQ), i–1 = k1’ CcQехр[(1–a)fQ)]. Q - доля поверхности, занятая промежуточным поверхностным комплексом МеВадс. Так как прямая и обратная реакции стадии (47) являются химическими, не связанными с переносом заряда через границу раздела фаз, то их скорости не зависят от потенциала электрода. Поэтому экспонента, включающая величину φ, в кинетических уравнениях отсутствует. Так как влияние (1–Q) и Q в предэкспоненциальном множителе кинетических уравнений невелико по сравнению с экспоненциальным членом, их можно считать константами, а уравнения упростить. Тогда имеем: i1 = k1 CA ехр(–bfQ), i–1 = k–1 CcQ ехр[(1–b)fQ)]. В случае квазиравновесия i1 = i–1, и, следовательно, k1 CA ехр(–bfQ) = k–1 Cc ехр[(1–b)fQ)]
ехр(fQ) = Для лимитирующей стадии (48) можно записать: i2 = k2’’ QCд ехр(bφF/RT) ехр(afQ) Примем для всех стадий a = b = 0,5, что, по существу, и использовано ранее, так как индексы у a и b были опущены. Тогда: i2 = k2’Cд ехр(bφF/RT) ехр(afQ) С учетом приведенной ранее зависимости Q от СА и Сс: i2 = k2’Cд ехр(bφF/RT) ехр или i2 = k2’Cд ехр(bφF/RT) ехр i2 = k2Cд ехр(bφF/RT) и окончательно имеем (опуская индексы) i2 = kCд Таким образом, использование изотермы Темкина легко позволяет интерпретировать дробные порядки по различным частицам (ионам, молекулам). Учитывая, что величина a может быть и не равной 0,5, порядок реакции также может быть несколько больше или меньше 0,5. Тафелевский наклон, однако, остается в rex же пределах, что и в случае использования изотерм Генри или Ленгмюра. Для разобранного случая: bа = Предлагаем читателю самостоятельно вывести кинетическое уравнение с использованием логарифмической изотермы (изотермы Темкина) для процесса, предложенного Дарвишем, Лоренцом, Хильбертом и Росвагом: Fe + С1– « Fe(Cl)aдc + е; Fe(Cl)aдc + H+ ® Fe(C1H+)aдc Fe(C1H+)aдc « FeCl+ + H+ +e, и для процесса, предложенного Чином и Ноубом: Fe + С1– + H2O « Fe(C1ОH)адс– + H+ +e, Fe(C1ОH)адс– ® FeClOH + e ; FeClOH + H+ « Fe2+ +С1–+Н2О. Предлагаем читателям себя проверить, сопоставив свои результаты с полученными в статье В.И.Вигдоровича и Л.Е.Цыганковой, опубликованной в журнале «Электрохимия». 1976. Т.12. №9. С. 1430 - 1436. В научной литературе впервые Вигдорович В.И., Цыганкова Л.Е. рассмотрели трехстадийные процессы ионизации металла с последней лимитирующей стадией с позиций описания адсорбции промежуточных продуктов посредством изотермы Темкина (Таблица 2). Это связано со значительными математическими трудностями. Если же их и пытались рассмотреть с этих позиций, то все исследователи для предшествующих лимитирующей квазиравновесных стадий использовали уравнения, не учитывающие функцию bfQ, то есть, по существу, сводили на нет саму идею. Таблица 2 Механизмы (I - III) анодного растворения железа

Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|

 ехр(bFφ/RT)+ k’’
ехр(bFφ/RT)+ k’’  ехр(bFφ/RT)
ехр(bFφ/RT) iа = k44
iа = k44 
 +
+ 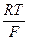 ln
ln  ;
; еφF/RT
еφF/RT ехр
ехр 
 ;
; - const и
- const и  = Ка/aH+
= Ка/aH+ ехр
ехр 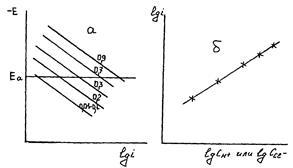
 +е, (45)
+е, (45)
 = k45
= k45  еφF/RT
еφF/RT получаем
получаем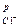 еφF/RT
еφF/RT C
C  еφF/RT,
еφF/RT,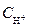 + k45в
+ k45в  )C
)C  ln
ln  ,
, ,
, ,
, = DG
= DG 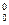 - fRTQ ,
- fRTQ ,
 (МеХ)адс +е
(МеХ)адс +е

 МеВадс + С; (47)
МеВадс + С; (47)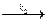 Ме+ + BД + е. (48)
Ме+ + BД + е. (48) = ехр[(1–b)fQ+bfQ];
= ехр[(1–b)fQ+bfQ];



 ехр(bφF/RT)
ехр(bφF/RT)