|
|
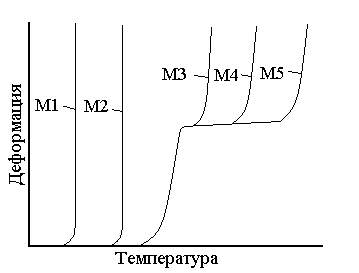
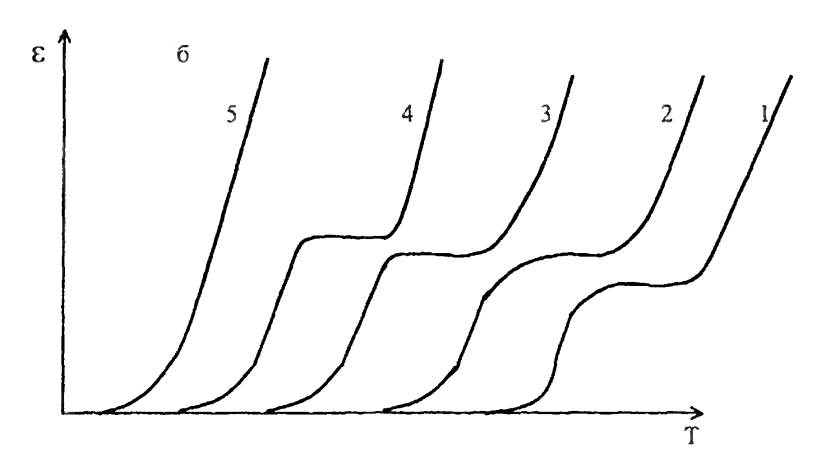
Влияние различных факторов на вид термомеханической кривой аморфных линейных полимеровХарактер и форма получаемых термомеханических кривых зависят от большого числа различных факторов, среди которых наибольшее значение имеют такие свойства полимера, как молекулярная масса, фазовое состояние, полимолекулярность и полярность полимеров, наличие в полимере пластификаторов, наполнителей, механическое напряжение и т.д. Влияние молекулярной массы полимера. На рис.5 видно, что для низших гомологов (кривые 1–2) не характерно высокоэластическое состояние. Однако с увеличением степени полимеризации для них наблюдается повышение Тс как следствие роста вязкости. Начиная с определенной молекулярной массы полимера, характерной только для данного полимергомологического ряда, появляется высокоэластическое состояние, охватывающее по мере роста молекулярной массы все больший интервал температур (кривые 3–5).
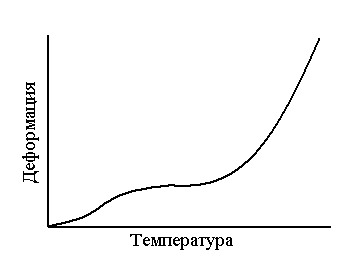
Рис. 5. Термомеханические кривые полимеров одного полимергомологического ряда (М5 >М4 >М3 >М2 >М1) Таким образом, начиная с некоторого члена полимергомологического ряда, с увеличением молекулярной массы наблюдается постоянство Тс и последовательное повышение Тт. Эта закономерность позволяет применить данные термомеханического анализа для определения молекулярных масс полимеров. [4,5,6] Величина молекулярной массы, начиная с которой появляется Тт, зависит от гибкости цепи: чем жестче цепь, тем выше молекулярная масса. Высокомолекулярные полимеры с гибкими цепями характеризуются низкими Тс и высокими Тт, т. е. с широким температурным интервалом эластичности (от –70 ºС до +200 ºС). Высокомолекулярные полимеры с более жесткими цепями имеют высокие Тс и небольшой интервал эластичности (от 100 ºС до 160 ºС). [5,6] Зависимость Тс полимеров от молекулярной массы позволяет оценить механический (кинетический) сегмент цепи. При определенной молекулярной массе полимера температура стеклования перестает зависеть от молекулярной массы и у полимергомологов появляются признаки высокоэластического состояния, т.е. проявляется гибкость цепи. У полимеров различного химического строения гибкость цепи проявляется при различных степенях полимеризации, например, у неполярных полимеров – при молекулярной массе порядка 1000, а у полярных – 12000. Очевидно, что длина цепи полимера, начиная с которой исчезает зависимость Тс от молекулярной массы и появляется разность между Тс и Тт, может служить мерой кинетической гибкости цепи. Такая длина совпадает с механическим сегментом полимера и характерезует его гибкость следующим образом: чем короче сегмент – тем больше гибкость. [4] Влияние полимолекулярности и полярности полимеров на температуру текучести. Для полимолекулярных полимеров термомеханическая кривая приобретает «размытый» характер, т.к. фракция полимера с различными молекулярными массами переходит в вязкотекучее состояние при неодинаковых температурах (рис. 6).
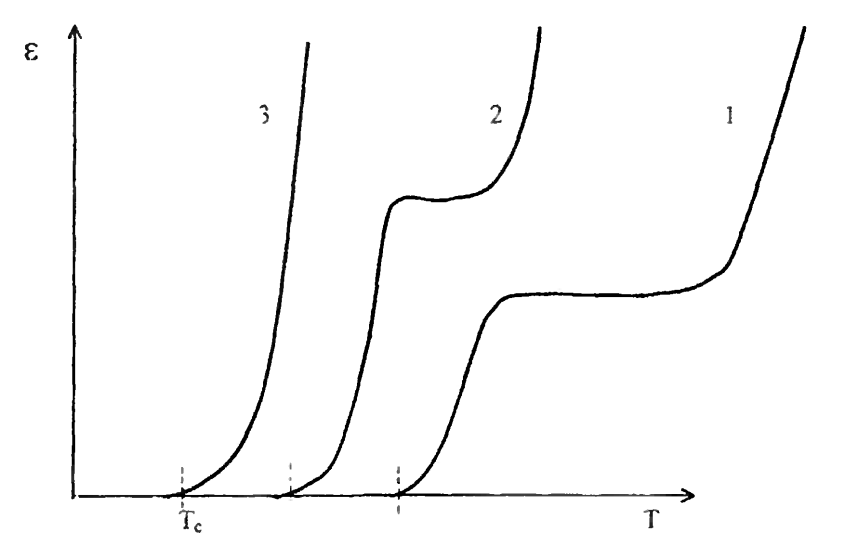
Рис. 6. Влияние полимолекулярности на термомеханические свойства полимера Полярность макромолекул также существенно влияет на Тт полимера. Поскольку взаимодействие между полярными цепями сильнее, чем между неполярными, вязкость полярных полимеров выше. Поэтому для того, чтобы вызвать перемещение цепей как единого целого, полярный полимер необходимо нагреть до более высокой температуры, т.е он обладает более высокой температурой текучести. [6] Влияние сшивки макромолекул. Сшитые аморфные полимеры при небольшом числе поперечных связей между макромолекулами характеризуются термомеханической кривой, приведенной на рис.7.
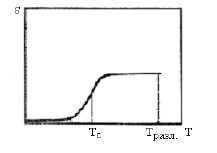
Рис. 7. Термомеханическая кривая сшитого аморфногополимера Узлы сетки препятствуют относительному перемещению центров тяжести полимерных цепей. В сшитом полимере вязкотекучее состояние отсутствует, и высокоэластическая деформация проявляется вплоть до температуры разложения (Тразл), выше которой термомеханическая кривая имеет сложный характер. При увеличении частоты пространственной сетки высокоэластическая деформация уменьшается, и, когда число поперечных связей превысит одну на каждый сегмент, трехмерный полимер будет деформироваться как обычное твердое тело, то есть высокоэластическая деформация перестанет проявляться.[1] Влияние пластификации. При внешней пластификации силы межмолекулярного взаимодействия между цепями уменьшаются. Появление промежуточного слоя (пластификатора между цепями) в полимере облегчает перемещение цепей, в результате чего всегда снижается температура стеклования и увеличивается пластичность (текучесть) полимера, что улучшает его переработку. [5] По мере увеличения количества пластификатора в полимере с гибкими цепями происходит полное вырождение высокоэластического состояния (рис.8). При этом переход полимера из стеклообразного происходит сразу в вязкотекучее состояние.
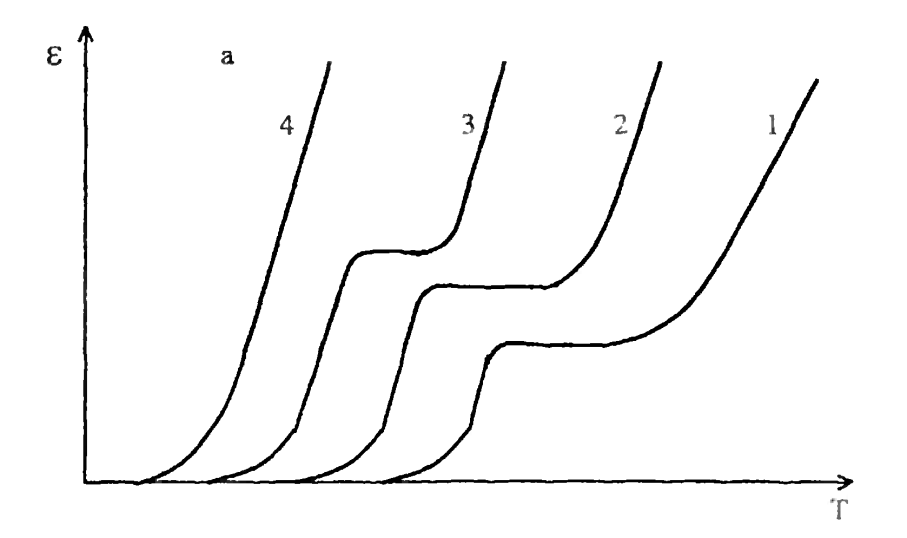
Рис.8. Влияние пластификатора на форму термомеханической кривой гибкоцепного полимера (содержание пластификатора растет с увеличением номера кривой) Для жесткоцепных полимеров со слабо выраженной высокоэластичностью (рис.9) при введении пластификатора вначале наблюдается расширение высокоэластического состояния (кривые 1-3).
Рис.9. Влияние пластификатора на форму термомеханической кривой жесткоцепного полимера (содержание пластификатора растет с увеличением номера кривой) Затем разность между температурой текучести и стеклования начинает убывать (кривая 4), после чего наблюдается переход к вязкотекучим смесям (кривая 5), застывающим в виде стекла без высокоэластического состояния. С практической точки зрения снижение температуры стеклования при введении пластификаторов позволяет расширить температурную область высокоэластического состояния полимеров, т.е. повысить морозостойкость. Понижение температуры текучести и вязкости полимерных расплавов позволяет существенно понизить температуру переработки, что особенно важно для полимеров с низкой термической стойкостью. [7] Влияние механического напряжения. Величина действующего напряжения является одним из основных факторов, определяющих форму термомеханической кривой. По своему влиянию на состояние полимера и уровень его деформации напряжение эквивалентно температуре. С ростом напряжения термомеханическая смещается в область низких температур. Площадка высокоэластичности проявляется только в определенном интервале напряжений (рис.10). При больших напряжениях полимер сразу переходит в вязкотекучее состояние (кривая 3), и область высокоэластичности вырождается.
Рис.10. Влияние напряжения на форму термомеханических кривых. Напряжение растет а увеличением номера кривой Температура стеклования практически линейно понижается с увеличением напряжения вплоть до температуры хрупкости, при которой сегментальная подвижность становиться невозможной, и образец хрупко разрушается. [7] Влияние введения наполнителей. Наполнители изменяют структуру полимера в результате сорбции сегментов макромолекул поверхностью наполнителя для образования связей полимер – наполнитель. Наполненные полимеры можно представить себе как двухфазную систему, состоящую из твердой фазы, расположенной около частиц наполнителя, и «мягкой фазы», на которую не распространяется влияние наполнителя, т.е. по существу она представляет собой не наполненный полимер. Для наполненных полимеров характерны две температуры стеклования, соответствующие стеклованию мягкой (Тс) и твердой фазы (Тс´). Как правило Тс´>Тс, так как в твердой фазе резко снижена сегментальная подвижность. Разность температур стеклования ∆Т = Тс´–Тс зависит от степени взаимодействия полимер – наполнитель, и часто вместо двух Тс наблюдается расширение интервала стеклования со смещением Тс в сторону более высоких температур. [8] 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|