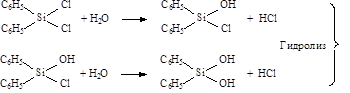|
|
Особенности реакций синтеза мономеров.Для этих реакций характерна большая глубина превращения, чем для реакции синтеза монофункционального соединения. Даже самая простая схема синтеза бифункционального соединения, например, нитрование бензола:
показывает, что при его получении достигается большая глубина превращения исходных соединений (бензола и азотной кислоты), чем при получении монофункциональных продуктов. И если в последнем случае можно получить продукт при малой глубине превращения (большой выход можно обеспечить возвратом в реакционную систему непрореагировавшего исходного сырья), то при синтезе бифункциональных соединений это невозможно. Большая глубина превращения определяет и особенности условий синтеза таких соединений: большую продолжительность процесса, более высокие температуры и концентрации, т.е. бифункциональные мономеры получают в более жестких условиях, чем монофункциональные соединения. Еще одной особенностью процессов получения бифункциональных соединений является их сложность. Практически все они состоят из ряда последовательно-параллельных реакций. Последовательность реакций получения полифункциональных соединений (моно-, би-, три- и т.д.) и их неразделенность в пространстве и времени приводят к тому, что реакционная смесь всегда содержит примеси соединений иной функциональности, т .е. в бифункциональном соединении всегда присутствуют примеси моно- или трифункциональных соединений. Это означает, что как в любой момент времени, так и в конце процесса в реакционной смеси находится набор различных продуктов. На рис. 6 показана зависимость содержания продуктов (моно-, ди-, три-и тетразамещенных) хлорирования дифенилолпропана от глубины процесса (содержания введенного хлора). Из рисунка видно, что на приведенных графиках нет ни одной точки, в которой содержание какого-либо из названных продуктов составляло 100%.
Рис. 6. Зависимость содержания различных продуктов от глубины превращения при хлорировании дифенилолпропана (Диана) в ядро): 1–монохлордиан, 2–дихлордиан, 3–трихлордиан, 4–тетрахлордиан, 5-диан. Набор продуктов при синтезе бифункциональных соединений становится еще более широким в случае протекания побочных реакций, т.е. реакций, не предусмотренных основным уравнением процесса. Так, при введении функциональной группы в ароматическое ядро практически всегда образуются три типа продуктов: орто-, мета- и паразамещенные. При протекании в одной и той же поликонденсационной системе реакций образования реакционных центров и реакций их превращения число побочных реакций возрастает. Для того чтобы уменьшить долю побочных реакций и, следовательно, количество примесных продуктов, при получении бифункциональных мономеров следует идти двумя путями: 1) повышать селективность (избирательность) целевого химического превращения; 2) проводить очистку мономеров. Повышение избирательности нужной химической реакции требует, по видимому, проведения значительной экспериментальной работы. Из общих соображений следует, что электрофильные реакции, как правило, менее селективны, чем нуклеофильные. При проведении реакции образования реакционных центров одновременно с побочными реакциями оптимальным является протекание их по разному кинетическому механизму. Довольно хорошо разработанным общим способом повышения выхода целевого продукта является метод защиты или метод защитных групп. Этот метод состоит в том, что после введения в соединение одной функциональной группы ее превращают в нереакционноспособную, далее вводят вторую группу (или проводят соответствующее химическое превращение), а затем снимают защитную группу. Например, при получении H2N–С6H4SO2CI (п-аминобензолсульфохлорида): прямое сульфохлорирование анилина (или обработка его хлористым сульфурилом) может привести к замещению аминогруппы остатком –SO2Cl. Поэтому анилин сначала обрабатывают муравьиной кислотой для образования соединения HCONH–С6H5, а затем хлорсульфоновой кислотой или хлористым сульфурилом для получения HCONHC6H4SO2CI, которое превращают в п-аминобензолсульфохлорид легким омылением, не затрагивающим группу CISO2. Интересным способом защиты функциональных групп является создание стерических препятствий путем введения рядом с функциональной группой большого объемного заместителя. Примером может служить получение бициклических фенолов окислительной поликонденсацией:
Для ароматических соединений кроме описанного способа прямой защиты можно использовать способ косвенной защиты, при котором вводимая защитная группа защищает не саму функциональную группу, а создает необходимые условия для протекания реакции только в одном положении, например:
Методы очистки, применяемые для удаления побочных продуктов из бифункционального мономера, не отличаются какими-либо особенностями (перегонка, дистилляция, возгонка, кристаллизация, экстракция и т. д. ). В случаях, когда эти традиционные методы очистки не дают существенного результата, мономер специально переводят в другое соединение, более легко очищаемое Так, пока не был разработан способ очистки терефталевой кислоты, синтез полиэтилентерефталата проводили через диметиловый эфир кислоты, который гораздо легче, чем кислота, подвергался очистке. Этот способ, естественно, экономически невыгоден. Однако в ряде случаев он может оказаться целесообразным, поскольку затраты на получение «промежуточного» мономера могут быть меньше убытков, которые приносит использование неочищенного продукта (плохое качество материалов и изделий, неустойчивость процессов их получения и т д). Образование реакционных центров в процессах Во многих случаях образование функциональных групп (реакционных центров) может происходить в поликонденсационной системе одновременно с самой поликонденсацией, то есть с образованием макромолекул. Это бывает в тех случаях, когда образовавшиеся реакционные центры очень активны, и поэтому предварительный их синтез и выделение невозможны. Так, например, в выше приведенном уравнении показано получение дифенилсиландиола. Синтез этого мономера не представляет значительных трудностей из-за его относительно низкой реакционной способности, обусловленной наличием в веществе стерически емких фенильных групп, препятствующих дальней его конденсации в олигомеры: nR2Si(OH)2 Однако вследствие высокой реакционной способности диорганосиландиолов с менее емкими заместителями многие из них синтезировать достаточно трудно с большим выходом. Некоторые соединения, такие как MeSi(OH)3 или VinSi(OH)3 не получены до сих пор. Однако, образуясь при гидролизе соответствующих органохлорсиланов эти мономеры дают начало химической сборке полиорганосилоксанов различного строения. В некоторых процессах выделение образовавшихся бифункциональных соединений из реакционной среды не всегда бывает целесообразным из чисто технологических соображений. Примером этого может служить синтез фенолформальдегидных смол. Другим ярким примером процесса поликонденсации со стадией образования реакционных центров во время поликонденсации является полирекомбинация. При полирекомбинации мономер, содержащий весьма подвижные атомы водорода (например, диизопропенилбензол), нагревают с определенным количеством перекиси, способной давать свободные радикалы. Процесс можно представить следующей схемой (см. ниже). На первой стадии реакционные центры образуются в результате отрыва подвижного атома водорода в молекуле мономера за счет взаимодействия его с активным радикалом R·, получившимся при распаде какой-либо перекиси:
На второй стадии происходит рекомбинация радикалов:
Далее следует отрыв атома водорода от образовавшихся димеров, рекомбинация получившихся макрорадикалов с образованием полимера строения:
Схема процесса полирекомбинации приведена на рис. 7. На основе приведенной упрощенной схемы полирекомбинации можно сделать следующий важный вывод относительно стадии образования реакционных центров при поликонденсационных процессах. Так же как и на других стадиях, на этой стадии может происходить выделение низкомолекулярного продукта реакции. Это еще раз говорит о том, что выделение низкомолекулярного продукта не является особенностью стадии образования макромолекулы при поликонденсации и, следовательно, не является спецификой таких процессов. Стадия образования реакционных центров предусматривает создание их как в молекулах исходного мономера, так и в молекулах олигомеров. Особенности этой стадии существенно сказываются на закономерностях поликонденсации, когда именно стадия образования реакционных центров является лимитирующей стадией всего процесса. Сюда следует отнести процессы, в которых выход основного продукта этой стадии - реакционных центров - мал из-за низкой скорости образования самих реакционных центров или из-за «невыгодной» константы соответствующего равновесия. Реакции образования реакционных центров в ходе самой поликонденсации весьма многообразны и аналогичны рассмотренным реакциям получения бифункциональных соединений со стабильными функциональными группами. Из изложенного следует принципиальная возможность получения поликонденсационных полимеров из такого доступного сырья, как углеводороды нефти, SO2, CO2 и т. д. Для этого необходима разработка эффективных методов управления достаточно длинной последовательностью реакций, составляющих стадию образования реакционных центров поликонденсационных процессов.
Рис. 7 а) Схема образования макромолекулы при полирекомбинации. Горизонтальные участки соответствуют образованию радикальных реакционных центров, наклонные–поликонденсации путем рекомбинации радикалов; б) Взаимосвязь между числом молекул в системе с их степенью поликонденсации: .А–С–поликонденсация удвоением, А–B–поликонденсация в реальных системах, х – степень превращения функциональных групп.. Природа мономера оказывает непосредственное влияние и на характер химических процессов, лежащих в основе реакции поликонденсации. В связи с этим различают равновесную и неравновесную поликонденсацию. Большинство используемых на практике поликонденсационных мономеров, содержат функциональные группы, взаимодействие которых приводит к выделению низкомолекулярного соединения. Если это вещество способно реагировать в процессе реакции с образовавшимся полимером, то процесс является равновесным. например:
Если образовавшееся при поликонденсации низкомолекулярное соединение не реагирует в условиях реакции с полимером, то поликонденсация является неравновесной, например:
При проведении неравновесной поликонденсации подбирают такие реагенты и условия, чтобы реакция протекала в области, далекой от равновесия, т.е. отсутствовали реакции деструкции полимера, обменные реакции и т.д. Это достигается проведением процесса при таких низких температурах, при которых обменные реакции замедлены, а исходные вещества достаточно реакционноспособны (например, дихлорангидриды дикарбоновых кислот). Для максимального превращения исходных веществ в полимер из реакционной системы необходимо удалять выделяющийся низкомолекулярный продукт реакции. С этой целью поликонденсацию проводят с одновременной отгонкой низкомолекулярного вещества при повышенных температурах (обычно в токе инертного, газа), причем на заключительной стадии процесс проводят в вакууме. Смещение равновесия путем удаления из сферы реакции низкомолекулярного соединения способствует получению полимера с высокой молекулярной массой. В некоторых случаях при взаимодействии функциональных групп параллельно поликонденсации может протекать реакция образования циклов. Возможность протекания циклизации или линейной поликонденсации определяется в основном строением исходного бифункционального вещества и условиями проведения реакции. Циклизация является основным направлением реакции в тех случаях, когда в результате образуются ненапряженные пяти- и шестичленные циклы (например, при циклизации аминомасляной, аминовалерьяновой и оксимасляной кислот). Если строение мономера таково, что при внутримолекулярном взаимодействии должны образоваться восьми-, девяти-, десятичленные циклы, то циклизации практически не происходит, и в результате образуются только линейные полимеры.
Глава 4 ЛАБОРАТОРНЫЕ РАБОТЫ Одним из широко применяемых в практике производства кремнийорганических полимеров является дифенилсиландиол. В настоящее время этот продукт используется и как мономер в синтезе различных кремнийорганических олигомеров и термоморозостойких каучуков, в качестве антиструктурирующего агента в каучуковых композициях и в синтезе душистых веществ. Дифенилсиландиол – (C6H5)2Si(OH)2 представляет собой кристаллическое вещество белого цвета, ограниченно растворимое в ацетоне, серном эфире, этаноле, не растворимое в воде, ароматических и алифатических углеводородах, четыреххлористом углероде. Температура плавления может быть разной в зависимости от кристаллической структуры и находится в широких пределах I28-I62оС, но интервал плавления конкретного чистого вещества (образца) должен быть не более 1оС. Химизм процесса, Дифенилсиландиол получают гидролизом дифенилдихлорсилана. С учетом возможным побочных реакций химическая схема процесса описывается схемой (см. след. стр.). На представленной схеме показаны реакции гидролиза Ph2SiCl2, по которым образуется целевой продукт – Ph2Si(OH)2 и побочные реакции: гомофункциональной конденсации (ГМФК), гетерофункциональной конденсации (ГТФК) и реакции циклизации. Для того, чтобы свести к минимуму побочные реакции необходимо знание механизмов этих стадий. Литературные данные и исследования этого процесса, проведенные на кафедре ХТЭОС, показывают, что ГМФК органосиланолов характеризуется порядками реакций по мономеру и катализатору больше единицы (по мономеру, как правило - второй порядок, по катализатору - первый). Эта реакция ускоряется в среде растворителей с высокой диэлектрической проницаемостью и любыми кислотными и основными катализаторами; зависимость скорости конденсации от температуры характеризуется достаточно большой энергией активации – порядка 15 - 20 ккал/моль. Поэтому, для уменьшения степени протекания ГМФК необходимо проводить синтез органосиланолов: 1) при низких концентрациях органохлорсиланов, 2) в среде растворителей с низкой диэлектрической проницаемостью, 3) в отсутствии в реакционной массе кислых и основных катализаторов, 4) при пониженных температурах (-5 ¸ -10оС). Схема гидролиза Ph2SiCl2:
Дальнейшие превращения:
Как показали исследования последних лет, из двух побочных реакций - ГМФК и ГТФК, наиболее значима последняя. Эта реакция протекает даже в условиях, когда берется значительный избыток воды по отношению к органохлорсилану. Одной из причин протекания ГТФК органохлорсиланов с продуктами их гидролиза - силанолами – является гетерофазный характер процесса гидролитической поликонденсации (ГПК) органохлорсиланов. Трехкомпонентная система органохлорсилан-вода-ацетон имеет одну пару взаимно нерастворимых компонентов (вода-органохлорсилан) и две пары взаимно растворимых компонентов (вода-ацетон и органохлорсилан-ацетон). Первой стадией процесса является массообмен компонентов реакционной системы, приводящий к разделению исходной гетерогенной смеси (Cji) на сосуществующие фазы, характеризующиеся недостатком воды по отношению к хлорсилану (органическая фаза Oi) и избытком (водная Wi). С позиций фазового равновесия можно дать простое качественное объяснение зависимости выхода силанолов от концентрации общего растворителя в реакционной системе, представленное на рис. 8. Для определенного содержания триорганохлорсилана в его смеси с водой–Хi, (рис. 8) рассмотрим тройной гетерогенный состав Cji, который разделяется на две сосуществующие фазы Oi и Wi. Любая водная фаза характеризуется молярным отношением воды к органохлорсилану m > 1,поэтому в них протекает полный гидролиз органохлорсилана с образованием силанола. В органических фазах возможны два интервала соотношения воды и органохлорсилана. При m ³ 1 в результате полного гидролиза образуется также в основном силанол. Если же фаза Oi характеризуется недостаточным количеством воды для полного гидролиза, то в ней протекает частичный гидролиз с последующей ГТФК неизрасходованного органохлорсилана с образовавшимся силанолом.
о Рис. 8 Фазовое равновесие системы органохлорсилан-вода-растворитель Кроме того необходимо учитывать, что реакция ГТФК должна протекать более быстро, чем ГМФК, так как при одинаковых нуклеофилах (молекулы силанолов), в первом случае субстратом является органохлорсилан с хорошей уходящей группой (Cl), а во втором - силанол – с плохой уходящей группой (OH). Поэтому выход силанолов будет зависеть от распределения воды и органохлорсилана по сосуществующим фазам. В связи с гетерофазным характером процесса ГПК органохлорсиланов необходимо иметь также ввиду, что для получения органосиланолов с высоким выходом, необходимо, чтобы процесс протекал в кинетической области, то есть при достаточно интенсивном перемешивании реакционной массы. Соблюдение выше указанных условий, а именно: проведение синтеза в среде растворителя с низкой диэлектрической проницаемостью, при интенсивном смешении компонентов реакционной системы и их оптимальном соотношении, а также при невысоких концентрациях органохлорсилана и пониженных температурах гарантирует высокий выход органосиланолов, и, в частности, дифенилсиландиола.
Схема реакции:
Таблица 6 Физико-химические свойства используемых веществ при 20оС
Порядок работы. Расчетная часть: в соответствии с уравнением реакции рассчитать количества реагентов и занести их значения в таблицу 7. Таблица 7
Проведение эксперимента. 1). Подготовка хроматографического анализа. Готовят батарейный стакан высотой 150-200 мм и выкладывают его внутреннюю поверхность чистой фильтровальной бумагой. Готовят элюент следующего состава: толуол - 35 мл и диоксан - 5 мл, который аккуратно заливают в батарейный стакан и закрывают стеклом. Включают и настраивают в соответствии с указаниями преподавателя TLC-сканер для количественной обработки хроматографических пластин. 2) В четырехгорлую круглодонную колбу, снабженную мешалкой, термометром, обратным холодильником и капельной воронкой, загружают 100 мл. ацетона, 4.Змл. воды и 25.2мл. эпихлоргидрина. Реакционную колбу со смесью помещают в водяную баню с теплой водой. При перемешивании содержимое колбы нагревают до 30-35оС. Затем из капельной воронки по каплям вводят 24,6 мл дифенилдихлорсилана с такой скоростью, чтобы температура реакционной смеси не поднималась выше 40°С. В течение всего синтеза контролируют рН среды по универсальному индикатору (рН должно быть равно 5 - 7). После ввода всего дифенилдихлорсилана содержимое колбы перемешивают в течение 10 мин. Проверяют рН. Анализируют состав гидролизата методом тонкослойной хроматографии. Затем реакционную смесь переносят в одногорлую колбу, добавляют 30 мл толуола и проводят отгонку ацетона на роторном испарителе в вакууме 250–240 мм. рт. ст. Осадок, белого цвета фильтруют и сушат на воронке Бюхнера или фильтре Шотта с использованием водоструйного насоса. Далее проводят дополнительную осушку продукта при 1-3 мм. рт. ст. Сухой продукт взвешивают и рассчитывают его выход. Качество дифенилсиландиола проверяют по присутствию в продукте примесей олигосилоксанов линейного и циклического строения методом количественной тонкослойной хроматографии, (см. ниже, где Ln º HO[Si(C6H5)2O]nH, Dn º [Si(C6H5)2O]n):
Задание 1. Изучить теоретическую часть; 2. Провести анализ дифенилсиландиола методом тонкослойной хроматографии; 3. Собрать экспериментальную установку синтеза Ph2Si(OH)2 ; 4. Подготовить анализ дифенилсиландиола методом ТСХ и определить температуру плавления; 5. Провести синтез дифенилсиландиола и определить выход продукта. Результаты оформить в лабораторном журнале. Лабораторная работа № 2. Схема реакции:
Повышение температуры синтеза приводит к тем же продуктам конденсации, которые указаны в лабораторной работе №1, что снижает выход дифенилсиландиола. Таблица 8 Физико-химические свойства используемых веществ при 20оС
Порядок работы. Расчетная часть: в соответствии с уравнением реакции рассчитать количества реагентов и занести их значения в таблицу 9.
Таблица 9
Проведение эксперимента. В четырехгорлую колбу, емкостью 500 мл, снабженную мешалкой, термометром, обратным холодильником и капельной воронкой, загружают 150 мл ацетона (или 200 мл этилацетата),1 мл воды дистиллированной,26 г бикарбоната натрия. Включают мешалку, и реакционную колбу нагревают на водяной бане до температуры 40 – 45оС и при этой температуре из капельной воронки начинают прикапывать 24.6 мл дифенилдихлорсилана, с такой скоростью, чтобы температура в колбе не поднималась выше 45оС. По окончании дозирования смесь перемешивают еще 5 мин и проверяют рН реакционной массы по универсальному индикатору. рН должна быть 7–7.5. Охлаждают реакционную колбу и фильтруют на воронке Бюхнера от осадка хлористого натрия и непрореагировавшего бикарбоната натрия, промывают осадок один раз 50 мл ацетона. Анализируют гидролизат методом тонкослойной хроматографии (ТСХ). Затем добавляют к гидролизату 30 мл толуола и проводят отгонку растворителя на роторном испарителе в вакууме 240 мм. рт. ст. и температуре водяной бани 45 – 50оС. При отгонке начинают выпадать кристаллы дифенилсиландиола. По окончании отгонки суспензию дифенилсиландиола охлаждают, и фильтруют и сушат на воронке Бюхнера или фильтре Шотта, с использованием водоструйного насоса. Далее проводят дополнительную осушку продукта при 1-3 мм. рт. ст. Сухой продукт взвешивают и рассчитывают его выход. Качество дифенилсиландиола проверяют по присутствию в продукте примесей олигосилоксанов линейного и циклического строения методом количественной тонкослойной хроматографии (см. ниже, где Ln º HO[Si(C6H5)2O]nH, Dn º [Si(C6H5)2O]n):
Задание 1. Изучить теоретическую часть; 2. Провести анализ дифенилсиландиола методом тонкослойной хроматографии; 3. Собрать экспериментальную установку синтеза дифенилсиландиола; 4. Подготовить анализ дифенилсиландиола методом ТСХ и определить температуру плавления; 5. Провести синтез дифенилсиландиола и определить выход продукта. Результаты оформить в лабораторном журнале. ЛИТЕРАТУРА 1. Мономеры для поликонденсации, под ред. Коршака В.В., М.: Мир, 1976. 2. Л. Б. Соколов Основы синтеза полимеров методом поликонденсации, М.: Химия, 1978, с. 5 - 42. 3. В.В. Киреев Высокомолекулярные соединения, М.: Высшая школа, 1992, с. 288-299. 4. П.В. Иванов Дисс. д.х.н., Теоретические основы гидролитической поликонденсации органохлорсиланов.
Иванов Павел Владимирович, Валентина Ивановна Маслова, Под общей редакцией доцента Иванова П.В. Мономеры для поликонденсации. Учебная разработка к дисциплине «Физика и химия высокомолекулярных соединений». Рецензенты – д.х.н., профессор Прокопов Н.И
Подписано в печать Печать офсетн., бум. офсетн., формат 60х90/16, тираж 50 экз., заказ 17 117571, Москва, пр. Вернадского, 86 Издательско-полиграфический центр МИТХТ
[i] Речь идет об основной стадии процесса – стадии образования макромолекулы. [ii] Буква "о" означает отсутствие функциональной группы данного типа. 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|



 HO(SiR2O)nH + (n-1)H2O
HO(SiR2O)nH + (n-1)H2O