|
|
Нарушения метаболизма фен.Нарушения метаболизма ГЛИ. Нетоксическая гиперглицинемия – причиной является дефицит ТГФК- редуктазы в реакции: ГЛИ + НАД + ТГФК à СО2 + метилен-ТГФК + НАДН2. Дефицит фермента или полное отсутствие в ткани мозга- является причиной гиперглицинемии. Существует четкая зависимость между остаточной активностью фермента и выживаемостью больных. У новорожденных отсутствует реакция на окружение, икота, гипотрофия и клонические судороги. У детей старшего возраста - задержка умственного развития и судороги. Содержание ГЛИ в моче выше нормы в 10-20 раз, а в крови в 3-5 раз при отсутствии накопления других аминокислот. Нарушения метаболизма ЦИС Наследственные нарушения обмена серосодержащих АмК.
Цистинурия- это аномалия обмена, связанная с образованием камней в почках, мочевом пузыре, мочеточниках. Как следствие отложение кристаллов цистина, на фоне глюкозурии, фосфатурии, общей аминоацидурии. Гомоцистинурия генетически гетерогенна. Полиморфизм проявляется в следующих формах: 1. Подвывиха хрусталика. Признаком является- эктопия хрусталиков- дрожание радужки при быстрых движениях головы ребенка. Нарушение связано с утолщением и фрагментацией зонулярных волокон, крепящих хрусталик к реснитчатому телу. Кроме того возможен остеопороз, вальгусное искривление голени, полая стопа. В половине случаев- УО, у 10-15% - судороги или тромбоэмболия. 2.Гетерогенная форма связанна с нарушением использования витамина В6. Гомоцистеин - продукт, лежащий на перекрестке всех превращений МЕТ. Соединяясь с СЕР- образует Цистатионин, или реметилируясь, он снова превращается в МЕТ. Дефицит цистатионинсинтетазы блокирует синтез цистатионина, и сопровождается усиленным превращением МЕТ в гомоцистеин. Последний тормозит образование поперечных сшивок в коллагене, блокирует активные группы ЛИЗ и окси-ЛИЗ, которые образуют поперечные сшивки. При отсутствии последних, с возрастом происходит накопление гомоцистеина, что можно расценивать как риск-фактор в развитии многих заболеваний. При гомоцистинурии в патологический процесс вовлекается множество органов и систем. Смерть наступает рано от тромбоза крупных артерий. При гомоцистинурии повышено выделение с мочой оксипролина и кислых ГАГ. В крови в 4-5 раз повышен МЕТ. В моче геперметеонинурия и гомоцистинурия. 3.Нарушение метаболизма фолиевой и ТГФК сопровождается мышечной адинамией. При отсутствии последних, с возрастом происходит накопление гомоцистеина, что можно расценивать как риск-фактор в развитии многих заболеваний.
Формы цистинурии
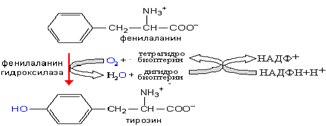
Нарушения метаболизма фен. Фенилкетонурия( болезнь Феллинга). Значительная часть ФЕН гидроксилируется при участии ФЕНалагидроксилазы и при участии биоптерина- донатора водорода. ( радикальный механизм с участием О2). Продуктом окисления является ТИР.
Нарушение основного пути катаболизма ФЕН проявляется гиперфеналанинемией и увеличением в крови и моче содержания фенилпирувата, фенилацецтата, фениллактата и фенилацетилглутамина. Известны две гетерогенные группы наследственных нарушений обмена Фен, сопровождающиеся фенилаланиннемией.: 1) Связанные с дефектом фенилаланингидроксилазы 2) Являющиеся последствием дефектов ферментов, участвующих в синтезе и метаболизме тетрагидробиоптерина. ФКУ- наследственное заболевание, причиной является –мутации в гене ФЕНАЛА-гидроксилазы. Частота ФКУ у новорожденных составляет 1: 10 000-1: 20 000. Тип наследования аутосомно- рецессивный Дефицит Тир и затрудненное его проникновение в ткань мозга через ГЭБ ( при избытке ФПВК), ограничивает синтез производных ТИР в нервной ткани (катехоламинов, серотонина). В патогенезе симтомов ФКУ основную роль играют нарушения обмена циклических аминокислот: - Увеличение конц. ФЕН ограничивает транспорт ТИР и ТРП через гематоэнцефалический барьер( ГЭБ) и тормозит синтез медиаторов: - избыток ФЕН нарушает ресинтез цереброзидсульфатов, участвующих в защите мозга от демиелинизации: - ФЕН и его производные – фенольные кислоты оказывают нейротоксическое действие. Они являются ингибиторами тирозингидроксилазы и триптофангидроксилазы -ключевых ферментов синтеза нейромедиаторов- L-дофа, норадреналина, серотонина. Коферментзависимая гиперфенилаланинемия.- это следствие мутации генов, контролирующих метаболизм тетрагидробиоптерина- дегидробиоптеринредуктазы и фенилаланингидроксилазы. Мутации в этих генах сопровождаются клиническими последствиями, близкими, но не совпадающими с ФКУ. Тетрагидробиоптерин необходим также для гидроксилирования ТРП и ТИР. Дефицит этого кофермента в равной степени нарушает метаболизм всех трех аминокислот.
Ранние симптомы ФКУ проявляются в виде состояния гиперреактивности, характерного запаха плесени, экземоподобной сыпи, а также отсутствии у детей, реакции на окружающих. В последующем появляется УО. Транзиторная тирозинемия новорожденных свойственна недоношенным детям Проявляется летаргией, снижением активности. Содержание Тир в крови повышено в 2-3 раза против нормы. Биохимически похожа на наследственную тирозинемию. Дефицит р- гидроксофенил- пируватгидроксилазы_ которая катализирует образование гомогентензиновой кислоты альтернативным путем. У недоношенных детей фермент тормозится Тир, содержащийся в пище. Дефицит аскорбиновой кислоты- усугубляет проявление недостаточности фермента. Алкаптонурия обусловлена с дефектом оксидазы гомогентензиновой кислоты. Недостаточность энзима наблюдается вскоре после рождения ребенка.. Клинически проявляется в зрелом возрасте, т.к. в тканях накапливается алкаптан- продукт полимеризации гомогентензиновой кислоты. 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|
 Цистинурия, цистиноз распространенное врожденное заболевание. Встречается с частотой ( 1: 600), уступая только ФПО. Нарушение метаболизма Метеонина связано со снижением или отсутствием активности фермента цистатионин- синтетазы
Цистинурия, цистиноз распространенное врожденное заболевание. Встречается с частотой ( 1: 600), уступая только ФПО. Нарушение метаболизма Метеонина связано со снижением или отсутствием активности фермента цистатионин- синтетазы