
|
|
Протокол N 8 от 30 августа 2006 г.К о м п л е к с н ы е С о е д и н е н и я
г. Ростов на Дону 2006г
Бикяшев Э.А.
УЧЕБНО-МЕТОДИЧЕСКОЕ ПОСОБИЕ
для студентов I-го курса дневного отделения биолого-почвенного факультета
К о м п л е к с н ы е С о е д и н е н и я
Печатается по решению кафедры общей и неорганической химии Ростовского государственного университета. Протокол N 8 от 30 августа 2006 г.
Рецензент - канд. хим. наук, ст. преп. Лисневская И.В. Ответственный редактор - д.хим.наук, проф.Лупейко Т.Г.
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ.
Среди многочисленных веществ имеется обширный класс соединений, состав которых может быть представлен как сочетание формул двух или большего числа хорошо известных соединений. Например, 2KJ∙HgJ2, AgCl∙2NH3, Cu(NO3)2∙4NH3, FeCl3∙6H2O, 3KCN∙Fe(CN)3. В конце XIXв уже было накоплено достаточно много экспериментальных данных о существовании подобных соединений высшего порядка, но известные на тот момент представления о строении веществ (теория химического сродства, валентности) не позволяли объяснить их состав, а тем более предсказать возможность целенаправленного синтеза подобных соединений, обосновать их свойства. В 1893г в немецком "Журнале неорганической химии" была опубликована статья Альфреда Вернера "О строении неорганических соединений", в которой впервые были изложены основы теоретических представлений о строении таких веществ, как кристаллогидраты, аммиакаты и двойные соли. А.Вернер отверг представления А.Кекуле о соединениях высшего порядка, как группах "склеившихся готовых" молекул, сохраняющих внутренний "распорядок" атомов. Изучая поведение аммиакатов и сложных галогенидов в растворах, он пришел к выводу, что между их составными частями происходит перегруппировка атомов и групп атомов. При этом зачастую меняется и характер связи: связи неионогенные (по которым не наблюдался распад на ионы раньше) становятся ионогенными и наоборот. Подобные соединения, содержащие сложные по-новому организованные многоатомные группировки, сохраняющие стабильность и в водных растворах, А.Вернер предложил называть комплексными (координационными) соединениями, в отличие от двойных солей. Например, в растворе алюмокалиевых квасцов с помощью качественных реакций легко можно обнаружить все ионы; один моль вещества (при написании формулы –K2SO4∙Al2(SO4)3∙24H2O) дает 8 моль ионов. Правда в растворе квасцов, также как и в растворе сульфата (и большинства других солей) алюминия обнаруживается кислая среда. Это свидетельствует об изменении свойств молекул воды под влиянием ионов Al3+. В пользу этого вывода говорит и то, что многие соли алюминия гигроскопичны, часто кристаллизуются из растворов в виде кристаллогидратов: Al2(SO4)3∙18H2O, Al(NO3)3∙9H2O, AlГ3∙6H2O (Г: Cl, Br, J). Эти особенности солей алюминия А.Вернеру поздней удалось объяснить на основе координационной теории и химической теории растворов Д.Менделеева, как результат сильной гидратации ионов Al3+ (образование прочных аквокомплексов). В то же время, в растворе 2KJ∙HgJ2 очень трудно выполнить качественные реакции на ионы J–: 2 KJ(р-р) + Pb(NO3)2 (р-р)
2KJ∙HgJ2 (р-р) + Pb(NO3)2 (р-р)
и Hg2+: Hg(NO3)2 (р-р) + КОН (р-р)
2KJ∙HgJ2 (р-р) + КОН (р-р)
Учитывая, что мольная электропроводность раствора KJ∙HgJ2 соответствует наличию в растворе трех моль ионов, можно предложить следующий вариант реакции диссоциации:
2KJ∙HgJ2 (р-р)
По словам А.Вернера "ионогенные связи K–J сменились на неионогенные Hg–J". Подобные эксперименты с другими соединениями высшего порядка позволили А.Вернеру сделать вывод о том, что атомы многих элементов (в основном металлических) после насыщения обычно присущих им валентностей способны проявить еще и дополнительную – координационную. Образованные за счет этого устойчивые в растворах сложные частицы составляют внутреннюю сферу соединения (комплекс). В его составе принято выделять центральный (обычно положительно заряженный) атом (комплексообразователь) и координированные вокруг него лиганды. Ионы, напрямую не связанные с центральным атомом, составляют внешнюю сферу комплекса и легко отделяются при растворении в воде. С учетом этих соображений приведенные выше формулы необходимо записывать следующим образом:
K2[HgJ4], [Ag(NH3)2]Cl, [Cu(NH3)4](NO3)2, [Fe(H2O)6]Cl3, K3[Fe(CN)6].
Аналогичные результаты были получены А.Вернером и при изучении аммиакатов.
В конце этого краткого обзора напомню, что речь идет о мыслях, идеях, высказывавшихся на рубеже XIX–XXвв, когда еще ничего не было известно о существовании электрона, не было никаких объективных сведений о структуре атома. Естественно, что все современные теории химической связи также были разработаны много позднее. Поэтому нет большого смысла подробно обсуждать взгляды А.Вернера на возможную природу "обычной" и "побочной" или "координационной" валентности. Но такие понятия, сформулированные в координационной теории А.Вернера, как комплексообразователь, лиганд, координационное число, внутренняя и внешняя сфера комплексов,пространственное строение комплексовактивно используются и в современной химии координационных соединений. За прошедшие годы настолько расширился круг известных (чаще синтезированных искусственно) веществ, настолько изменилось наше понимание об их строении, что стало довольно трудно провести четкую разграничительную линию между соединениями "комплексными" и более "традиционными". Нередко оказывается, что вещества, чьи формулы записываются очень просто и лаконично (Al2O3, ZnS, CoCl2), устроены сложней, чем тем соединения, которые принято называть комплексными (K2[PtCl6], [Co(NH3)6]Cl3). Поэтому в рамках данного изучаемого вами курса комплексными соединениямибудут называться вещества ионного или молекулярного строения, сохраняющие в водных растворах сложные ковалентно-связанные многоатомные группировки, центральные атомы, в которых, тем не менее, способны обратимо обменивать своих "соседей" (лиганды). · Чаще всего в качестве комплексообразователя выступают атомы (ионы) d–металлов (например, Fe, Fe2+, Fe3+; Cu+, Cu2+; Ni, Ni2+, Pd2+, Pt2+, Pt4+ и т.п.) ионы р–элементов больших периодов (например, Te4+; Bi3+, Sb3+, Sb5+; Sn2+, Sn4+, Ge4+ и т.п.), а также ионы s–элементов малых периодов (например, Be2+). Даже простое перечисление потенциальных комплексообразователей позволяет заметить, что эта способность наиболее характерна для d–элементов, а наименее характерно комплексообразование для s–элементов. Объясняется это тем, что связь комплексообразователь––лиганд не должна быть ни сильно ионной (иначе такие группировки будут полностью диссоциировать в водном растворе), ни сильно ковалентной (иначе такие группировки будут очень стабильны к замене лигандов, как, например, ионы SO42–). Внутренняя сфера таких соединений должна быть образована смешанным ионно-ковалентным типом связывания. Поэтому центральные атомы должны оказывать среднее поляризующее действие на свое окружение (лиганды).
В некоторых редких случаях к комплексным относят частицы, в составе которых центральным атомом выступает анион (отрицательно поляризованный атом): N–III в [NH4]+ –ион аммония, O–II в [OH3]+ – ион гидроксония, J–I в [JJn]––полииодид-ионы. · В качестве лигандов обычно выступают простые одноатомные анионы (Cl–, Br–, J–), двухатомные анионы (ОН–, SH–, СN–), некоторые многоатомные анионы, не имеющие в своем составе собственного многозарядного центрального атома (SCN–, NO2–, очень редко NO3–; SO32–, S2O32–; очень редко SO42–), а также полярные (NH3, H2O, CO) и легко поляризуемые молекулы (J2). Как уже отмечалось, связь комплексообразователь––лиганд должна быть в заметной степени ковалентной, а значит должна быть образована за счет обобществления электронов. Потенциальные комплексообразователи на своих валентных орбиталях порой вообще не имеют электронов (например, Be2+, Al3+, Ti4+, V5+), но пустые валентные орбитали у них у всех имеются. Значит, все они могут выступать акцепторами электронных пар. Потенциальные лиганды должны выступать в качестве доноров электронных пар.
Атомы азота в составе лигандов выступают только в качестве σ–доноров (устанавливают только σ–связь за счет собственной электронной пары); атомы фтора и кислорода являются и σ–донорами, и π–донорами (особенно хорошо связываются с комплексообразователями, имеющими много пустых валентных орбиталей); атомы серы ведут себя как σ–доноры, и π–акцепторы (за счет собственных пустых 3d–орбиталей). Но особенно сильными π–акцепторнымисвойствами обладают лиганды, связывающиеся атомами углерода (CN–и CO), т.к. в соответствии с современными теориями химической связи (метод молекулярных орбиталей) в указанных частицах на атомах углерода имеются более низколежащие по энергии (чем у атомов S) пустые орбитали. Соответственно цианидные комплексы более характерны для d–металлов, находящихся во второй половине соответствующих 3d–, 4d– 5d–рядов. Все приведенные выше лиганды могут установить только одну σ–связь с комплексообразователем, "занимают лишь одно место в его координационной сфере". Такие лиганды называют монодентантными. Крупные анионы многоосновных органических кислот содержат несколько атомов, способных выступить в качестве σ–доноров. Такие лиганды называются полидентантными(например, анион щавелевой кислоты С2О42–– оксалат ион, анион этилен-диамин-тетрауксусной кислоты (ЭДТА: ( –OOCCH2)2N–CH2-CH2–N(CH2COO– )2.
Пространственное строение октаэдрического комплекса с участием аниона этилен-диамин-тетрауксусной кислоты
Комплексы с их участием содержат циклические группировки и называются хелатными. Такие комплексы особенно стабильны к замене лигандов. Приведем некоторые примеры комплексных соединений с целью их систематизации в дальнейшем: [Fe(OH2)6]Cl3, K3[Fe(CN)6], K4[Fe(CN)6], [Fe(CO)5], K[FeCl4], K3[Fe(C2O4)3], K3[Fe(ЭДТА)]; H[AuCl4], K[Au(CN)2], K[Au(CN)4]; K2[HgJ4], [Ag(NH3)2]OH, Na[Ag(S2O3)2], Na[Ag(CN)2]; [Co(NH3)6]SO4, [Co(NH3)6]Cl3, [Co2(CO)8], [Co(OH2)6]SO4; [Al(OH2)6]Cl3, K3[Al(OH)6], K[AlCl4], K3[AlF6]; K[BF4], K[B(OH)4]; Число σ–связей, образуемых центральным атомом (число молекул или однозарядных ионов, удерживаемых во внутренней сфере комплекса) называется координационным числом комплексообразователя. Наиболее часто встречающиеся К.Ч. равны 6, 4 и у малозарядных ионов, замыкающих d–ряды, – 2. Геометрическая форма окружения центрального атома в этих случаях:
Варианты классификации комплексных соединений:
К особой группе придется отнести комплексы, где в качестве лигандов выступают молекулы СО.
3. По природе лигандов комплексы классифицируют на акво (L: H2O), аммиакаты (L: NH3), гидроксо (L: OH–), ацидо (L: анионы кислотных остатков). Среди ацидокомплексов следует различать хлорокомплексы (хлоридные), иодо (иодидные), циано (цианидные, L: CN–), родано (роданидные, L: SCN–) и т.п. Особые группы составляют карбонилы (L: CO), хелатные комплексы (L: C2O42–, ЭДТА и т.п.), комплексы с несколькими разными лигандами.
Номенклатура комплексов.
В соответствии с правилами Международного союза по теоретической и прикладной химии (ИЮПАК) наименования комплексных соединений образуются следующим образом. Независимо от заряда координационной сферы сначала называется анион, а затем – катион, как это делается в названиях простых солей. В названии координационной сферы (вне зависимости от того, заряжена она или нет) перечисляют все ее составные части справа налево, при этом сначала называют число лигандов, затем сами лиганды и, наконец, комплексообразователь с указанием его степени окисления. Для обозначения числа лигандов используют греческие числительные: 1 – моно, 2 – ди, 3 – три, 4 – тетра, 5 – пента, 6 – гекса, 7 – гепта, 8 – окта (приставка моно часто опускается). Названия некоторых распространенных лигандов приведены в таблице. Если координационная сфера заряжена положительно, то комплексообразователь называют по-русски в родительном падеже. Если координационная сфера имеет нулевой заряд, то комплексообразователь называют так же по-русски, но в именительном падеже. Если координационная сфера заряжена отрицательно, то комплексообразователь называют по-латыни, при этом латинское окончание –ум отбрасывают и заменяют на окончание –ат, характерное для анионов. Степень окисления комплексообразователя указывают в скобках римскими цифрами.
Например, рассмотренные в предыдущем разделе комплексные соединения называются так: K4[Fe(CN)6] – гексацианоферрат (II) калия K3[Fe(CN)6] – гексацианоферрат (III) калия [Al(OH2)6]Cl3 – хлорид гексаакваалюминия [Fe(CO)5] – пентакарбонилжелезо [Ag(NH3)2]Cl – хлорид диамин-серебра (I) [Pt(NH3)4Cl2]Cl2 – хлорид дихлоро-тетраммин-платины (IV) (NH4)2[Pt(OH)2Cl4] – дигидроксотетрахлоро-(IV)платинат аммония [Cr(H2O)3F3] – трифторо-триаква-хром [Co(NO2)2C(NH3)3] – динитро-хлоро-триаммин-кобальт
Названия комплексных соединений более сложного состава образуют, использую дополнительные правила: - если координационная сфера содержит разные лиганды, то сначала называют отрицательно заряженные лиганды (анионы), потом – электронейтральные лиганды (молекулы). - если название лиганда содержит греческие числительные, то число лигандов указывают так: 2 – бис, 3 – трис, 4 – тетракис.
Поведение комплексов в водных растворах.
Отношение комплексов к воде определяется их строением. Нейтральные комплексы молекулярного строения ([Pt(NH3) 2Cl4], [Fe(CO)5], [Co2(CO)8]) являются либо жидкостями, либо легкоплавкими кристаллическими веществами в зависимости от размеров молекул (энергии межмолекулярного дисперсионного взаимодействия). Все очень плохо растворимы в воде. Заметно лучше смешиваются с неполярными органическими жидкостями (бензол С6Н6, четыреххлористый углерод СCl4). Большинство других веществ представляют собой ионные кристаллы, в структуре которых регулярно чередуются разноименно заряженные ионы. Среди таких комплексов имеется немало хорошо растворимых в воде веществ. В процессе растворения молекулы воды сначала вступают в ориентационное взаимодействие с ионами, затем наступает этап донорно-акцепторного связывания (гидратация). Гидратированные ионы покидают кристалл и за счет диффузии равномерно распределяются в объеме раствора (см. химическую теорию растворения твердых веществ в воде): [Ni(NH3)6]SO4 (тв)
K2[HgJ4] (тв)
Процесс растворения таких соединений совпадает с их диссоциацией. Часто ее называют первичной диссоциацией комплексов. В то же время в полученных растворах не удается выполнить известные качественные реакции на ионы Ni2+ и Hg2+ (см. стр.1). Это говорит о высокой стабильности внутренней сферы. Однако устойчивость комплексного иона не является абсолютной. Такой вывод можно сделать, если использовать более чувствительные реактивы на ионы‑комплексообразователи. Ионы Ni2+ и Hg2+ имеют 3d8 и 5d10 конфигурации, поэтому могут выступать не только акцепторами электронных пар от лигандов при σ–связывании, но и сами могут предоставлять свои d–электроны для дополнительного π–связывания. Таким образом, эти ионы способны хорошо связываться с атомами (лигандами) σ‑донорами и π–акцепторами, например с ионами CN–, S2–:
ПРNi(CN)2= 3.0∙10-23 ПРNiS = 3.2∙10-19
ПРHg(CN)2= 3.0∙10–35 ПРHgS = 4.0∙10–53.
Поэтому при добавлении раствора Na2S в обоих случаях выпадают черные осадки сульфидов. Эти реакции свидетельствуют о том, что внутренняя сфера комплексов все же частично диссоциирует (вторичная диссоциация). Упрощенно соответствующие уравнения можно записать в виде:
[Ni(NH3)6]2+
[HgJ4]2–
На самом деле необходимо иметь в виду, что при слабо идущей диссоциации лиганды должны отщепляться последовательно, постадийно:
Константы соответствующих равновесий можно найти в справочной литературе. Их называют частными (ступенчатыми)константами нестойкостикомплексов:
К' = 0.9 К" = 0.18 К''' = 6.5∙10–2 К' = 6.0∙10–3 К" = 1.7∙10–4
К'''' = 1.8∙10–2 К''''' = 5.8∙10–3 К'''''' = 1.7∙10–3 К''' = 1.1∙10–11 К'''' = 4.5∙10–14
Но, строго говоря, эти уравнения также не вполне точны, т.к. в соответствии с ними получается, что КЧ центрального атома постепенно уменьшается и он может в водном растворе полностью "утратить'' свое окружение. На самом же деле в итоге получаются гидратированные ионы, поэтому сам процесс диссоциации следует понимать как последовательную замену соответствующих лигандов на молекулы воды (обсуждать "процесс диссоциации аквокомплексов" в водном растворе вообще бессмысленно):
Однако надо признать, что выглядят эти уравнения довольно громоздко, поэтому записываются в таком виде очень редко. Но именно анализ подобных равновесий позволяет объединить в единую систему разные, на первый взгляд, обменные равновесия в растворах комплексов: замена лигандов, осаждение и растворение осадков, гидролиз, кислотно–основные реакции. Позднее мы немного коснемся этих вопросов. Вернемся к упрощенным записям уравнений диссоциации внутренней сферы. Промежуточные стадии этих равновесий представляют интерес достаточно редко, но тогда важно понимать, что константа суммарного равновесия представляет собой произведение ступенчатых констант нестойкости:
[Ni(NH3)6]2+
Знание соответствующих констант позволяет прогнозировать возможность (полноту) протекания различных реакций в растворах комплексов. Как известно, равновесия реакций обмена в растворах электролитов смещены в сторону более прочного связывания ионов (в осадки или слабые электролиты, в том числе, в прочные внутренние сферы комплексов). Скажем тот факт, что в растворе тетраиодомеркурата калия трудно получить осадки оксида ртути (II) и иодида свинца легко объяснить на основе анализа следующих равновесий:
K2[HgJ4] + 2 КОН
Сокращенное ионное уравнение имеет вид:
[HgJ4]2– + 2 ОН–
Кнест = 5∙10–31 ПР(HgO) = 3∙10–12 Kw = 1∙10–14
Кнест = 5∙10–31 ПР(HgO) ∙Kw =3∙10–26 Как видно, в комплексе связывание ионов более прочное, чем в продуктах, поэтому образование осадка очень маловероятно. Аналогично выглядит ситуация и при попытке осадить иодид свинца[1]:
K2[HgJ4] + 2 Pb(NO3)2
[HgJ4]2– + 2 Pb2+
Кнест = 5∙10–31 ПР(PbJ2) = 8∙10–9
Кнест = 5∙10–31 ПР(PbJ2) ∙ ПР(PbJ2) = 6.4∙10–17
В растворе сульфата гексаамминникеля (II) не удается получить осадок карбоната никеля:
[Ni(NH3)6]SO4 + Na2CO3
или в ионном виде: [Ni(NH3)6]2+ + CO32–
Кнест = 1.9∙10–9 ПР(NiCO3) = 1.3∙10–7
А вот сульфиды никеля (II) и ртути (II) гораздо менее растворимы и осаждаются даже в растворах комплексов. При этом внутренняя сфера центрального атома полностью разрушается:
Итак, эти реакции подтверждают, что внутренняя сфера не является абсолютно устойчивой. Если этот вывод усвоен, то полезно задуматься над вопросом: что представляют собой полученные осадки? Насколько просто устроены вещества с таким простым составом? Насколько данные реакции отличаются от ранее приводившихся примеров реакций замены лигандов? Так вот, оба сульфида являются ионно-ковалентными (для d–металлов характерно такое сочетание типов связей) полимерами с высокими КЧ.

Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|
 PbJ2↓ + 2 KNO3 (р-р)
PbJ2↓ + 2 KNO3 (р-р) нет признаков реакции
нет признаков реакции Уже в 40-е годы ХХв, когда были разработаны методы структурного исследования, было показано, что зеленый празеохлорид содержит ионы хлора в транс-положении, а фиолетовый виолеохлорид представляет собой цис–изомер.
– Сколько пространственных изомеров следует ожидать среди соединений состава CoCl3∙3NH3? Как выглядит окружение атома кобальта в каждом случае? Как меняется электропроводность растворов одинаковой молярной концентрации в ряду:
CoCl3∙6NH3 – CoCl3∙5NH3 – CoCl3∙4NH3 – CoCl3∙3NH3?
Какое координационное число имеют ионы Co3+в этих комплексах?
Уже в 40-е годы ХХв, когда были разработаны методы структурного исследования, было показано, что зеленый празеохлорид содержит ионы хлора в транс-положении, а фиолетовый виолеохлорид представляет собой цис–изомер.
– Сколько пространственных изомеров следует ожидать среди соединений состава CoCl3∙3NH3? Как выглядит окружение атома кобальта в каждом случае? Как меняется электропроводность растворов одинаковой молярной концентрации в ряду:
CoCl3∙6NH3 – CoCl3∙5NH3 – CoCl3∙4NH3 – CoCl3∙3NH3?
Какое координационное число имеют ионы Co3+в этих комплексах?
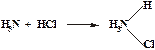
 [NH4]Cl
[NH4]Cl 

 связываются за счет атомов O, S, C;
связываются за счет атомов O, S, C;
 связывается за счет электронных пар атомов S или N;
связывается за счет электронных пар атомов S или N;
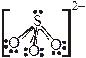
 связывается, как правило, атомом S; связывается концевым атомом S;
связывается, как правило, атомом S; связывается концевым атомом S;


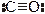 связывается атомом С
связывается атомом С
 в качестве лигандов выступают крайне редко, т.к. электронные пары атомов кислорода активно участвуют в πp–d–связывании с собственным центральным атомом.
в качестве лигандов выступают крайне редко, т.к. электронные пары атомов кислорода активно участвуют в πp–d–связывании с собственным центральным атомом.

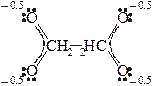

 [Ni(NH3)6]2+ (р-р) + SO42–(р-р);
[Ni(NH3)6]2+ (р-р) + SO42–(р-р); Ni2+ + 6 NH3
Ni2+ + 6 NH3 [Ni(NH3)5]2+ + NH3
[Ni(NH3)5]2+
[Ni(NH3)5]2+ + NH3
[Ni(NH3)5]2+  [Ni(NH3)4]2+ + NH3
[Ni(NH3)4]2+
[Ni(NH3)4]2+ + NH3
[Ni(NH3)4]2+  [Ni(NH3)3]2+ + NH3
[Ni(NH3)3]2+
[Ni(NH3)3]2+ + NH3
[Ni(NH3)3]2+ 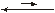 [Ni(NH3)2]2+ + NH3
[Ni(NH3)2]2+
[Ni(NH3)2]2+ + NH3
[Ni(NH3)2]2+ 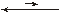 [Ni(NH3)]2+ + NH3
[Ni(NH3)]2+
[Ni(NH3)]2+ + NH3
[Ni(NH3)]2+  Ni2+ + NH3
Ni2+ + NH3
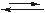 [HgJ3]– + J–
[HgJ3]–
[HgJ3]– + J–
[HgJ3]–  [HgJ2] + J–
[HgJ2]
[HgJ2] + J–
[HgJ2]  [HgJ]+ + J–
[HgJ]+
[HgJ]+ + J–
[HgJ]+  Hg2+ + J–
Hg2+ + J–


 HgО↓ + Н2О + 4 КJ
HgО↓ + Н2О + 4 КJ 2 PbJ2↓ + 2 KNO3 + Hg(NO3)2
2 PbJ2↓ + 2 KNO3 + Hg(NO3)2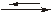 NiS ↓ + 6 NH3 + Na2SO4
[Ni(NH3)6]2+ + S2–
NiS ↓ + 6 NH3 + Na2SO4
[Ni(NH3)6]2+ + S2–  HgS↓ + 2 KNO3 [2]
[HgJ4]2– + S2–
HgS↓ + 2 KNO3 [2]
[HgJ4]2– + S2–