
|
|
Расчет изменения энтропии сложного процессаИзменение энтропии сложного процесса, состоящего из нескольких стадий, равняется сумме изменений энтропий каждой отдельной стадии этого процесса:
Например, изменение энтропии при обратимом превращении 10,0 г льда, находящегося при -10 °С, в пар при 120 °С, если С(Н2О, ТВ) = 2,03 Дж г-1 К-1; DНпл. льда = 331,89 Дж г-1; С(Н2О, Ж) = 4,18 Дж г-1 К-1; DНисп. Н2О = 2253,4 Дж г-1. С(Н2О, Г) = 1,93 Дж г-1 К-1; Вычисляют следующим образом. Данный процесс представим из пяти стадий: 1. Нагревание льда от -10 °С до 0 °С, DS1. 2. Плавление льда, DS2. 3. Нагревание жидкой воды от 0°С до 100 °С, DS3. 4. Испарение воды, DS4. 5. Нагревание пара от 100 °С до 120 °С, DS5. Изменение энтропии в 1, 3, 5 стадиях рассчитывают по уравнению
В стадиях 2, 4 – по уравнению Общее изменение энтропии составит: 4.4. Расчет изменения энтропии Изменение энтропии химического процесса равно алгебраической сумме стандартных энтропий участников реакции с учетом их стехиометрических коэффициентов, причем энтропии продуктов реакции берутся со знаком плюс, а энтропия исходных веществ – со знаком минус. Для реакции, протекающей по уравнению aА + bB ® mM + nN
В общем виде:
(с учетом стехиометрических коэффициентов). 4.5. Критерии направления протекания процессов По изменению энтропии можно судить о направлении и пределах протекания процессов только в изолированных системах. В случае закрытых и открытых систем необходимо также учитывать изменение энтропии окружающей среды. Решение последней задачи сложно или невозможно. Поэтому в термодинамике для изучения открытых или закрытых систем используют другие термодинамические функции, так называемые термодинамические потенциалы, изменение которых позволяет определить направление процессов и пределы их протекания без учета изменений в окружающей среде. В частности, к термодинамическим потенциалам относится функция состояния, называемая энергией Гиббса, которую обозначают через G. Понятие об энергии Гиббса было введено на основе объединенного уравнения первого и второго начал термодинамики. Это уравнение может быть выведено следующим образом. Из первого начала термодинамики следует:
из второго начала термодинамики для обратимого процесса получаем:
Для необратимого процесса:
Подставляя значение для обратимого процесса
для необратимого процесса
Уравнение (4.14) называют объединенным уравнением первого и второго начал термодинамики для обратимых процессов. Так как внутренняя энергия и энтропияявляются функциями состояния, то их изменения не зависят от того, как протекает данный процесс, обратимо или необратимо, следовательно:
т.е. работа, совершаемая при обратимом процессе, больше работы, производимой системой при необратимом процессе при условии, что начальное и конечное состояния системы одинаковы в обоих случаях. Имея в виду, что работа, производимая системой при обратимом процессе, является максимальной для данного изменения состояния системы, преобразуем уравнение (4.14):
группируя величины с одинаковыми индексами, получим:
так как Если система кроме полезной работы совершает работу против силы внешнего давления (Р = const), то для обратимого процесса
или
где
Для необратимого процесса
Учитывая, что
получим
Группируя величины с одинаковыми индексами, находим:
Величину ( Раньше эту функцию состояния называли изобарно-изотермическим потенциалом. Таким образом, G = Имея в виду последнее уравнение, можно записать:
или, так как
Из уравнения (4.25) следует, что максимальная полезная работа, совершаемая системой в обратимом изобарно-изотермическом процессе, равна уменьшению энергии Гиббса. Для необратимого процесса путем аналогичного преобразования справедливо
т.е. уменьшение энергии Гиббса в необратимом процессе больше производимой системой работы (полезной). Зная, что
уравнение (4.24) запишем следующим образом:
или
Последнее уравнение может быть представлено следующим образом:
или
Из этого следует, что изменение внутренней энергии системы можно представить как сумму трех слагаемых:
«Связанная энергия» тем больше, чем больше энтропия данной системы. Таким образом, энтропию можно рассматривать как меру «связанной энергии». Определим изменение энергии Гиббса при протекании обратимого и необратимого процессов. Для этого продифференцируем уравнение (4.27), помня при этом, что дифференциал произведения двух функций равен сумме производных, взятых в предположении, что в данных условиях изменяется только одна из функций, а другая остается постоянной. В результате дифференцирования получим:
Из соотношений (4.13) и (4.15) следует:
где знак равенства относится к обратимым, а знак неравенства – к необратимым процессам. Предполагая, что никакой работы, кроме работы расширения, не производится, получаем:
Подставляя найденное значение
Из уравнения (4.34) следует, что для обратимых процессов при постоянных Р и T и при условии, что из всех видов работы может производиться только работа расширения,
так как
Поскольку обратимые процессы являются равновесными, т.е. такими, когда система в каждый момент времени находится в состоянии равновесия, то уравнение (4.35) является критерием равновесия. Протекание необратимых процессов, к которым относятся все самопроизвольные процессы, сопровождается уменьшением энергии Гиббса (Р и Т постоянны), так как при этом
Поэтому неравенство (4.36) является критерием направления самопроизвольных процессов. Из неравенства (4.36) следует, что при переходе системы из неравновесного состояния в равновесное G уменьшается и в момент равновесия достигает минимального значения: Gравн. = Gmin. (4.37) Уравнение (4.37) представляет собой второй критерий достижения равновесия в открытых и закрытых системах. Лекция 5. 5.1. Понятие о химическом потенциале Химическую реакцию можно рассматривать с точки зрения изменения в системе количеств продуктов реакции и исходных веществ. Изменение энергии системы с увеличением числа молей данного вещества на единицу времени при неизменных прочих параметрах системы можно рассмотреть на следующей простой модели. В сосуде при постоянном давлении и температуре находятся три газа: азот n(N2), кислород n(О2) и водород n(H2) в одинаковых количествах: n(N2) = n(O2) = n(H2). Из сосуда можно вывести или (ввести) один моль какого-либо газа, например кислорода. Очевидно, для этого нужно затратить энергию:
Изменение энергии Гиббса системы в этом случае может быть записано как:
где mi – коэффициент пропорциональности между изменением энергии и количества i-го компонента. Определим изменение энергии Гиббса системы от изменения давления. Учтем, что
Так как изменение энергии системы рассматривается при постоянной Т, следовательно -SDТ = 0, а так как dn = 1 (количество газа в системе изменяется на 1 моль), то
Так как V = const, следовательно, изменение mi в системе пропорционально изменению энергии Гиббса системы. Из этого следует, что коэффициент пропорциональности mi характеризует изменение энергии Гиббса системы, связанной с изменением количества вещества. При достижении системой равновесия dG = 0 и, следовательно,
Для одного моля газа (идеального) V = RT/Р. Подставим это выражение в уравнение (5.5) и проведем интегрирование. Получим:
где m1 и m2 – химический потенциал чистого газа при давлениях Р1 и Р2 (соответственно) и температуре Т соответственно:
Удобно выбрать Р1, равным 1 атм, тогда m1 можно обозначить как m0, т.е. химический потенциал одного моля чистого газа при 1 атм и температуре Т (стандартный химический потенциал), тогда приведенные уравнения могут быть записаны так: m = m0 + RT lnP, (5.9) G = G0 +RT lnP. (5.10) С другой стороны, можно рассматривать любой вид энергии как произведение двух величин: фактора интенсивности и фактора емкости.
Интенсивная величина (фактор интенсивности), показывающая изменение энергии системы с изменением массы, носит название химического потенциала. 5.2. Экзергонические и эндергонические реакции Величина DG служит мерой способности системы производить работу. Она позволяет решить вопрос, может ли реакция протекать самопроизвольно. Реакциия протекает самопроизвольно только в том случае, если происходит уменьшение энергии Гиббса системы. Такие реакции называют экзергоническими. Если же энергия Гиббса системы возрастает, то для осуществления реакции необходимо затратить работу. Такие реакции называют эндергоническими. В живых организмах реакцию, которая в данных условиях не является самопроизвольной, поскольку протекание ее связано с увеличением «свободной энергии», можно осуществить путем сопряжения ее с другой реакцией, характеризующейся достаточно большой отрицательной величиной изменения энергии Гиббса. Условием такого сопряжения будет наличие интермедиата, т.е. общего для обеих реакций вещества. Пример: 1. А + В « С + Д; DG1o > 0. 2. Д + К « М + Г; DG2o < 0. 3. Ф + В + К « С + М + Г; DG3 < 0. Для живых организмов можно привести много примеров сопряженных реакций. Особенно большое значение имеют реакции гидролиза таких соединений, как аденозинтрифосфат (АТФ), аденозиндифосфат (АДФ), аргининфосфат, креатинфосфат, характеризующихся величинами изменения энергии Гиббса от –29,99 до –550,21 кДж/моль. Отметим, что широко распространенный термин «богатая энергией связь» в этом случае в химическом понимании неточен, поскольку в данной интерпретации он относится не к энергии связи в химическом понимании, а к стандартной энергии Гиббса, характеризующей гидролиз фосфатного соединения. Энергия связи в химическом понимании – это энергия, которая должна быть затрачена для разрыва связи в молекуле с образованием нейтральных атомов или радикалов. Концепция, согласно которой энергия локализована в богатой фосфатной связи и освобождается при ее расщеплении, получила значительное распространение. Однако энергия разрыва связи и, следовательно, представление о том, что процесс расщепления связи поставляет энергию и что энергия запасается в связи, не имеет физического смысла. Большая величина стандартной энергии Гиббса гидролиза фосфатных соединений связана с тремя факторами. Первый – более высокая термодинамическая стабильность продуктов гидролиза по сравнению с исходными соединениями. Второй – в исходных соединениях имеются близко расположенные отрицательно заряженные кислотные группы и часть энергии, освобождаемой при гидролизе, связана с электростатическим отталкиванием этих групп в исходном состоянии. Третий – это вклад энергии ионизации групп, образующихся при гидролизе. Таким образом, совокупность протекающих при гидролизе процессов определяет общий, конечный эффект. 5.3. Взаимосвязь энтальпийного и энтропийного Из уравнения DGo = DНo – ТDSo (5.11) видно, что знак DG зависит от двух факторов: DH – энтальпийного фактора, характеризующего стремление системы к минимуму полной энергии системы и DS – энтропийного фактора, характеризующего стремление системы к переходу в более беспорядочное состояние. Значит DG является интегральным показателем, отражающим взаимосвязь энтальпийного и энтропийного факторов. Рассмотрим несколько возможных вариантов: 1) DНo< 0; DS > 0; DG< 0 – это относится к экзотермическим реакциям, идущим с увеличением энтропии. Такие реакции протекают самопроизвольно в стандартных условиях. 2) DНo > 0; DS < 0; DG > 0 –эндотермические реакции, идущие с уменьшением энтропии. Такие реакции возможны в сопряжении с другими процессами; 6Н2О + 6СО2 ® С6Н12О6. 3) Другие возможные варианты: DН < 0; DS < 0; |DН| > |TDS|; DGo< 0; |DН| < |TDS|; DGo > 0; |DН| = |TDS|; DGo = 0. 5.4. Понятие о равновесии химической реакции. Учение о химическом равновесии является одним из важнейших разделов физической химии. Начало ему положено работами французского ученого К. Бертолле (1799 г.) и в наиболее общем виде развито норвежскими учеными К. Гульдбергом и П. Вааге (1867 г.), установившими закон действующих масс. Химическое равновесие устанавливается в системах, в которых протекают обратимые химические реакции. Обратимой химической реакцией называют такую реакцию, продукты которой, взаимодействуя между собой в тех же условиях, при которых они получены, образуют некоторые количества исходных веществ. С эмпирической точки зрения химическим равновесием называют состояние обратимой химической реакции, при котором концентрации реагирующих веществ в данных условиях не меняются со временем. Примером здесь являются реакция получения йодоводорода из водорода и йода Н2 (Г) + J2 (Г) « 2НJ (Г), и реакция этерификации С2Н5ОН (Ж) + СН3СООН (Ж) « С2Н5СООСН3 (Ж) + Н2О (Ж), так как образующиеся продукты реакции (йодоводород и уксусно-этиловый эфир) способны в тех же условиях, при которых они получены, образовывать исходные вещества. Необратимой химической реакцией называют такую реакцию, продукты которой не взаимодействуют друг с другом при тех же условиях, в которых они получены, с образованием химических веществ. Пример – реакция разложения бертолетовой соли на кислород и хлорид калия: 2KClO3 (Т) ® 2KCl (Т) + 3О2 (Г). Образующиеся в этих случаях продукты реакции не способны взаимодействовать друг с другом с образованием исходных веществ в условиях их получения. Понятия об обратимых и необратимых химических реакциях не следует путать с понятиями об обратимых и необратимых процессах (в том числе и химических реакций) в термодинамическом смысле. 5.5. Уравнение изотермы химической реакции Рассмотрим обратимую химическую реакцию, протекающую между газообразными веществами в изобарно-изотермических условиях в соответствии с уравнением аА + вВ « mM + nN. Изменение количеств реагирующих веществ в процессе протекания химической реакции до достижения равновесия можно рассматривать как изъятие из системы веществ или добавление продуктов реакции. Изменение химических потенциалов каждого компонента в этом случае выразится следующим образом: mА = amоА + a RTln CA, mB = bmоB + b RTln CB, mM = mmоM + mRTln CM, mN = nmоN + nRTln CN. Общее изменение химического потенциала в системе при протекании химической реакции можно выразить как Dm = Snim(прод) - Snim(исх) (5.12) или Dm = mmоM + RT ln Обозначим mmоM + nmоN - amоА- bmоB через Dmо и, преобразовав сумму логарифмов в логарифм дроби, получим:
Необходимо отметить, что через С обозначены исходные концентрации участников реакции. При установлении химического равновесия изменение химического потенциала равно нулю. В соответствии с этим уравнение (5.13) примет вид:
при этом через Из уравнения (5.14) следует, что
при постоянной температуре – величина постоянная, так как Dmо есть величина постоянная для данной реакции и, следовательно,
Уравнение (5.15) является математическим выражением закона действующих масс, установленного в 1867 году норвежскими учеными К. Гульдбергом и П. Вааге, согласно которому отношение произведения равновесных концентраций продуктов реакции, возведенных в степени, показатели которых равны их стехиометрическим коэффициентам, к произведению равновесных концентраций исходных веществ в степенях, показатели которых равны их стехиометрическим коэффициентам, для данной обратимой реакции есть величина постоянная при данной температуре. Эта величина получила название константы химического равновесия. Принимая во внимание, что изменение энергии Гиббса системы DG=SmiDn, аналогичными рассуждениями можно показать, что
Приведенное уравнение называют уравнением изотермы химической реакции, или уравнением изотермы Вант-Гоффа. Оно позволяет производить расчет изменения энергии Гиббса при протекании химической реакции. Условия достижения системой состояния равновесия определяются DG= 0, следовательно, можно записать для состояния равновесия: DGо = -RTlnK. (5.17) Подставив значение DGо в уравнение (5.16), получим:
Критерием самопроизвольного протекания химического процесса является уменьшение свободной энергии (энергии Гиббса) системы, т.е. DG <0. Анализ уравнения (5.18) показывает, что это возможно в том случае, если
В этом случае система, находящаяся в неравновесном состоянии, будет стремиться к состоянию равновесия, и при этом концентрация исходных веществ будет уменьшаться, а продуктов – увеличиваться, т.е. реакция будет протекать в прямом направлении. Рассмотрим условие, при котором
Если DG > 0, то, следовательно, самопроизвольное протекание прямой реакции невозможно. Однако при этом протекание обратной реакции должно сопровождаться уменьшением энергии Гиббса, т.е. DG<0 и, следовательно, при условии Уравнение (5.17) можно использовать для расчета констант химического равновесия. Для этого необходимо вычислить изменение стандартной энергии Гиббса, происходящее в результате протекания реакции при постоянных Р и Т. Их расчет приводит по уравнению DGo = Sni DGo(пр)- Sni DGo(исх). (5.19) Зная DGo, вычисляют константу химического равновесия:
5.6. Зависимость константы химического равновесия Для установления зависимости константы химического равновесия от температуры рассмотрим состояние равновесия химической реакции при произвольных Т1 и Т2. Предположим, что DНo и DSo в интервале температур от Т1 до Т2 остаются постоянными, тогда константы химического равновесия при Т1 и Т2 могут быть выражены: DG1o = - RT lnK1 DG1o = DНo –T1DSo; DG2o = - RT lnK2 и соответственно DG2o = DНo –T2DSo. Решим систему приведенных уравнений относительно константы равновесия химической реакции:
Вычтем из первого уравнения второе:
или
(интегральная форма уравнения изобары). Полученная зависимость позволяет определить смещение состояния равновесия при изменении температуры. Предположим, что DHo>0 – эндотермическая реакция, тогда при увеличении температуры (Т2>Т1) величина ln K1/K2< 0, что выполняется при условии K1< K2 (K1/K2< 1), т.е. равновесие химической реакции смещается в сторону образования продуктов реакции. Уменьшение температуры (Т2<Т1) приведет соответственно к смещению равновесия в сторону образования исходных веществ. При условии, что реакция – экзотермическая (DHo < 0), повышение температуры (Т2 >Т1) определяет Часто для анализа направления смещения равновесия химической реакции при изменении температуры удобно пользоваться дифференциальной формой уравнения изобары 1) DH< 0; 2) DH > 0; dT > 0 – для эндотермической реакции увеличение температуры ведет к увеличению K; 3) DТ< 0 – для экзотермической реакции уменьшение t ведет к уменьшению К химического равновесия. Проведенный анализ зависимостей константы химического равновесия показывает, что смещение системы из состояния равновесия путем внешнего воздействия (изменение температуры, давления, концентрации) приводит к увеличению энергии Гиббса, и самопроизвольно могут протекать те процессы, которые противодействуют возрастанию DG, т.е. процессы, связанные с уменьшением энергии Гиббса, что является термодинамическим обоснованием принципа Ле-Шателье. Принцип смещения равновесия химической реакции, сформулированный Ле-Шателье, а затем Брауном, можно выразить следующим образом: если на систему, находящуюся в устойчивом равновесии, подействовать извне, изменив какое-нибудь из условий, определяющих равновесие, то в системе усилится то из направлений процесса, течение которого ослабляет влияние произведенного воздействия, и положение сместится в том же направлении. Лекция 6. 6.1. Взаимосвязь химического потенциала С точки зрения термодинамики живые клетки представляют собой открытые системы. Как указывалось ранее, открытой называется система, которая обменивается с внешней средой материей и энергией. Основополагающие термодинамические уравнения, написанные для изолированных и закрытых систем, должны быть в этом случае модифицированы. В них необходимо ввести члены, отражающие изменения массы системы. В этом случае дифференцирование уравнения для термодинамических функций U, Н и G будут иметь следующий вид:
где m – химический потенциал, n – число молей соответствующего компонента. Каждому компоненту системы в уравнении соответствует свой член mdn. Если в правой части каждого уравнения все переменные, за исключением n1, будут оставаться постоянными, то мы получим следующий ряд равенств:
а также из уравнения (6.1)
Из приведенных выше уравнений видно, что в зависимости от того, какие величины остаются постоянными, химический потенциал представляет собой парциальную молярную внутреннюю энергию, энтальпию или свободную энергию. Поскольку химические процессы в живых клетках и в большинстве экспериментальных систем in vitro протекают при постоянной температуре и при постоянном давлении, наиболее полезным является определение химического потенциала как парциальной молярной свободной энергии. При обсуждении свободной энергии Гиббса мы подчеркиваем значение этой величины как критерия равновесия в системах при постоянных Т и Р: равновесие достигается при DG = 0. Следовательно, когда свободная энергия перестает в системе меняться, химический потенциал любого компонента системы остается постоянным. Таким образом, условие равновесия может быть записано как DmI = 0 при Р и Т = const. Аналогично тому, как мы определяли стандартные изменения для других термодинамических функций и получали стандартные ΔН°,ΔS°и ΔG°, мы можем определить также стандартный химический потенциал μ°как изменение свободной энергии на 1 моль вещества, образующегося, расходующегося или переходящего из одной фазы в другую в своем стандартном состоянии (т.е. при давлении в 1 атм, при определенной температуре и в стандартной эталонной форме). 6.2. летучесть Стремление к улетучиванию. При описании поведения гетерогенной фазы (например, жидкости, контактирующей с газовой фазой, или твердого вещества в присутствии своих паров) очень часто пользуются понятием летучесть. Летучесть определяется притяжением между молекулами. Другими факторами, определяющими летучесть, являются кинетическая энергия молекул вещества, его форма, размер и т.д. Наиболее удобным параметром, отражающим летучесть, является давление пара данного вещества. Если в газовой фазе присутствует более чем одно вещество, равновесные давления каждого из них называют парциальными давлениями пара в соответствии с определениями, данными законом парциальных давлений Дальтона. 6.3. Идеальный раствор Как реальные газы при определенных температурах и давлениях по своим свойствам близки идеальным газам, так и реальные растворы при определенных условиях разбавления приближаются к идеальному раствору. Идеальным является такой раствор, в котором свойства, присущие растворителю и растворенному веществу, не меняются из-за присутствия новых компонентов, если не считать возможных изменений этих свойств при разбавлении. Стремление к улетучиванию для молекул растворителя или растворенного вещества уменьшается только в той мере, в которой наличие молекул другого компонента пространственно затрудняет или вообще делает невозможным уход молекул из раствора. В случае идеальных растворов объем является аддитивным, а температура при смешивании не изменяется. В реальных растворах растворитель и растворенное вещество взаимодействуют друг с другом. Например, если спирт обладает низкой летучестью из-за сильного диполь-дипольного взаимодействия, то при смешивании его с бензолом вследствие разделения диполей полярные молекулы спирта легче переходят в газовую фазу, и парциальное давление его пара будет выше. Возможно и наоборот в случае, например, растворения неполярного вещества в полярном растворителе. Для того, чтобы применять законы для идеальных растворов в случае реальных, необходимо введение некоторых ограничений. Один из таких принципов носит название предельного закона. Он гласит, что если содержание растворенных молекул в растворе относительно мало, то взаимодействие между ними будет минимальным и, следовательно, они будут слабо влиять на поведение растворителя. Таким образом, законы идеальных растворов можно применять к реальным растворам, если речь идет о разбавленных. Для рассмотрения свойств реальных растворов при относительно высоких концентрациях растворенного вещества имеются также уравнения, однако они достаточно сложны. Прежде чем приступить к обсуждению некоторых относительно простых уравнений, тем не менее, позволяющих рассчитывать давление пара, точку замерзания, точку кипения, а также осмотическое давление растворов, познакомимся с двумя фундаментальными соотношениями, лежащими в основе предельных уравнений, – с правилом фаз и уравнением Клаузиуса-Клапейрона. Правило фаз. На рис. 6.1 показана фазовая диаграмма, количественно описывающая поведение гетерогенной системы, содержащей гомогенные области-фазы. Фазы отделены друг от друга заметными границами, которые называют поверхностями раздела. Каждая фаза гомогенна, однако она не непрерывна. Число фаз равно числу отделенных друг от друга структур, существующих в различных физических состояниях в системе. В одной системе могут существовать несколько жидких или твердых фаз, например: вода – масло, лед – твердая соль. Различают как число фаз, так и число компонентов, образующих фазу.Под числом компонентов понимают число независимых химических индивидуальностей. На рис. 6.1 показана хорошо известная фазовая диаграмма воды. Она является однокомпонентной. Если в воде растворить соль, то получим двухкомпонентную систему. Если растворить два белка в буфере, получим пятикомпонентную систему – два белка, вода, соль и кислота. Изучение гетерогенных систем сильно упростилось после предложенного в 1874 году Гиббсом обобщения, которое получило название «правило фаз». Оно состоит из очень простого соотношения: а = с – р + 2, где с – число компонентов системы, р – число фаз и f – число степеней свободы системы, т.е. число независимых переменных – таких, как температура, давление и концентрация, которые необходимы для полного описания системы. 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2026 stydopedia.ru Все материалы защищены законодательством РФ.
|
 . (4.8)
. (4.8) .
. .
. .
. . (4.9)
. (4.9) (4.10)
(4.10)
 ; (4.11)
; (4.11)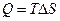 . (4.12)
. (4.12) <
<  . (4.13)
. (4.13) ; (4.14)
; (4.14) <
<  . (4.15)
. (4.15)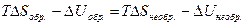 и
и  >
>  . (4.17)
. (4.17)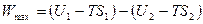 , (4.18)
, (4.18) и
и  – функции состояния, то величина (
– функции состояния, то величина (  должна быть также функцией состояния.
должна быть также функцией состояния. (4.19)
(4.19) ,
, – максимальная полезная работа, совершаемая системой в обратимом изобарно-изотермическом процессе. Из уравнения (4.18) получаем для обратимого процесса:
– максимальная полезная работа, совершаемая системой в обратимом изобарно-изотермическом процессе. Из уравнения (4.18) получаем для обратимого процесса: . (4.20)
. (4.20) =
=  . (4.21)
. (4.21) ,
, . (4.22)
. (4.22) . (4.23)
. (4.23)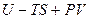 ), которая является функцией состояния, так как
), которая является функцией состояния, так как 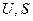 и
и 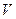 есть функции состояния, называют энергией Гиббса и обозначают G.
есть функции состояния, называют энергией Гиббса и обозначают G. ,
, ;
;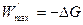 . (4.25)
. (4.25) <
< 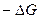 , (4.26)
, (4.26) ,
, (4.27)
(4.27) . (4.28)
. (4.28) (4.29)
(4.29) . (4.30)
. (4.30) –часть внутренней энергии, способная при изобарно-изотермических условиях превращаться в работу;
–часть внутренней энергии, способная при изобарно-изотермических условиях превращаться в работу; –часть внутренней энергии, затрачиваемая системой на совершение работы против сил внешнего давления;
–часть внутренней энергии, затрачиваемая системой на совершение работы против сил внешнего давления; . (4.31)
. (4.31) , (4.32)
, (4.32) . (4.33)
. (4.33) в уравнение (4.31) и зная, что
в уравнение (4.31) и зная, что 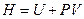 , следовательно
, следовательно  , имеем:
, имеем: . (4.34)
. (4.34) ,
, и
и  =0. (4.35)
=0. (4.35) < 0. (4.36)
< 0. (4.36) при Р и Т, n(N2) и n(H2) = const. (5.1)
при Р и Т, n(N2) и n(H2) = const. (5.1) , (5.2)
, (5.2)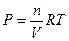 , поэтому это фактически равнозначно изменению энергии Гиббса, связанному с изменением концентрации вещества в системе:
, поэтому это фактически равнозначно изменению энергии Гиббса, связанному с изменением концентрации вещества в системе: . (5.3)
. (5.3) . (5.4)
. (5.4)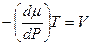 . (5.5)
. (5.5) , (5.6)
, (5.6) , (5.7)
, (5.7) . (5.8)
. (5.8) + nmоN + RTln
+ nmоN + RTln 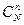 - amоА - RTln
- amоА - RTln  - bmоB – RTln
- bmоB – RTln  .
. . (5.13)
. (5.13) , (5.14)
, (5.14) обозначены концентрации участников реакции при достижении системой равновесия.
обозначены концентрации участников реакции при достижении системой равновесия.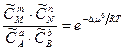
 . (5.15)
. (5.15)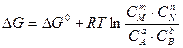 . (5.16)
. (5.16) . (5.18)
. (5.18) .
. (т.е. DG >0).
(т.е. DG >0). . (5.20)
. (5.20) (1),
(1), (2).
(2).
 (5.21)
(5.21) , из чего следует, что K1>K2 (K1/K2>1), т.е. равновесие экзотермической реакции при повышении температуры смещается в сторону образования исходных участников реакции, и соответственно, понижение температуры проведения реакции – в сторону продуктов.
, из чего следует, что K1>K2 (K1/K2>1), т.е. равновесие экзотермической реакции при повышении температуры смещается в сторону образования исходных участников реакции, и соответственно, понижение температуры проведения реакции – в сторону продуктов. .
.
 dT > 0 – для экзотермической реакции увеличение температуры приводит к уменьшению константы химического равновесия, если dT < 0 уменьшение t приводит к росту K (константы химического равновесия);
dT > 0 – для экзотермической реакции увеличение температуры приводит к уменьшению константы химического равновесия, если dT < 0 уменьшение t приводит к росту K (константы химического равновесия); (6.1)
(6.1) (6.2)
(6.2)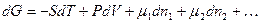 (6.3)
(6.3)
