
|
|
Термохимическая теория растворения электролитовПервой стадией образования раствора является разрыв связей в молекулах или кристаллах растворяемого вещества. Особенно большие затраты энергии требуются на разрушение кристаллической решетки. Энергией кристаллической решетки называют тепловой эффект гипотетического процесса изохорно-изотермического перехода 1-го моля вещества в стандартном состоянии из кристаллов в состояние ионного газа. Выберем в качестве примера для рассмотрения хлорид магния. Энергия решетки - это изменение внутренней энергии в следующем процессе:
Для расчета термохимической энергии кристаллической решетки используют приведенный ниже цикл Борна – Габера. Ho, кДж/моль ½¾¾¾Mg2+(g) + 2Cl-(g)¾¾¾¾¾¾¾¾¾ ½ ½ ½DfHoИОНОВ ½ ½ ½ ½ ½ ½¾¾¾Mg(s)+Сl2(g)¾¾¾¾¾¾¾–––¾¾ ½DНoРЕШ ½ ½ ½ ½ ½DfHo(MgCl2) ½ ½ ½ ½ ½ ¯ ½ ½¾¾¾¾MgCl2(s)¾¾¾¾¾¾¾¾¾¾¾¾ Как показывает расчет, затраты энергии на разрыв связей в кристалле настолько велики, что процесс растворения был бы невозможен при отсутствии гидратации. Одновременно с разрушением кристаллической решетки протекает гидратация ионов молекулами воды. Гидратация обусловлена ион-дипольными взаимодействиями в случае катионов щелочных металлов, водородными связями в случае анионов и образованием аквокомплексов многозарядных катионов по донорно-акцепторному механизму. Энергия образующихся в процессе гидратации связей компенсирует энергию кристаллической решетки, что делает возможным растворение электролитов. Энергией гидратации называют тепловой эффект изохорно-изотермического процесса перевода одного моля ионов из состояния ионного газа в стандартный раствор. То есть это изменение внутренней энергии в следующем процессе:
Аналогично для хлорид-иона вычисляем энергию гидратации +69 кДж/моль. Эндоэффект гидратации обусловлен, по-видимому, разрывом водородных связей в структуре воды при внедрении в неё аниона хлора, который сам водородные связи не образует. Термохимические энергии гидратации изменяются в соответствии со следующими закономерностями: Энергии гидратации катионов при равном заряде в несколько раз больше по абсолютной величине, чем энергии гидратации анионов. Например, энергия гидратации 2-зарядного сульфид-иона равна -475 кДж/моль. Энергия гидратации катиона калия равна -262 кДж/моль, значительно отрицательнее, чем у аниона хлора с теми же зарядом и радиусом. Причина в более высокой плотности заряда на отрицательном полюсе молекул воды (положительный заряд разделен между двумя атомами водорода) и в дополняющем простое ион-дипольное взаимодействие донорно-акцепторном связывании неподеленных электронных пар кислорода с вакантными орбиталями катионов. Энергии гидратации приближенно пропорциональны ионному потенциалу, равному z/r. Рост экзоэффекта гидратации с увеличением заряда иона является определяющим фактором и виден из приведенных примеров при переходе от K+ к Mg2+ или от Cl- к S2-. Снижение экзоэффекта гидратации с ростом радиуса иона демонстрирует переход от Mg2+ к Са2+, для которого энергия гидратации равна всего лишь 1369 кДж/моль. Энергия гидратации растет по модулю при переходе от катионов главных подгрупп Периодической системы к катионам переходных металлов. Например, радиусы Mg2+ и Zn2+ равны соответственно 0,74 Å и 0,83 Å. Согласно радиусам, экзоэффект гидратации должен быть у катиона цинка ниже, однако его энергия гидратации равна -2883 кДж/моль. Причина в том, что катионы переходных металлов образуют в растворе аквокомплексы с донорно-акцепторными связями. Тепловой эффект растворения соли равен сумме энергии кристаллической решетки и энергий гидратации составляющих её ионов. Это положение поясняет приведенный ниже термохимический цикл. Ho, кДж/моль ½¾¾¾Mg2+(g) + 2Cl-(g)¾¾¾¾¾¾¾¾¾¾¾¾ ½ ½ ½ ½ ½ ½ ½DHoРЕШ ½ ½ ½ ½ ½¾¾¾MgCl2(s)¾¾¾¾¾¾ ½DHoГИДР ½ ½ ½ ½ ½DsolHo ½ ½ ¯ ¯ ½¾¾¾Mg2+(aq) + 2Cl-(aq)¾¾¾¾¾¾¾¾¾¾¾
Благодаря большому экзоэффекту гидратации катионов магния с малым радиусом и большим зарядом, растворение хлорида магния протекает с выделением тепла. Следует отметить, что термохимическая теория гидратации построена на экспериментальных данных о тепловых эффектах рассматриваемых процессов и является более корректной по сравнению с расчетными теориями. Теория гидратации Борна Недостатком термохимической теории является невозможность разделить суммарную энергию гидратации катиона и аниона, составляющих электролит, на вклады в неё отдельных ионов. Такое разделение в термохимии проводят, принимая, что энтальпия образования гидратированного катиона водорода в стандартном состоянии равна нулю, однако данное допущение формально и не соответствует действительности. Поэтому представляет интерес теоретический расчет энергии гидратации ионов, предпринятый Борном (1920 г.). Борн исходил из модели «заряженный шарик в пространстве». Шарик создает электрическое поле. Потенциал электрического поля на расстоянии r равен e/r/ Для поля в воде шара с радиусом r, толщиной dr, объемом
где ri – радиус иона. Поскольку работа равновесного процесса равна изменению энергии Гиббса. При переходе 1 моль ионов из вакуума в среду с диэлектрической проницаемостью ε выделится энергия, равная
Данное уравнение получило название уравнения Борна.
Дифференцированием этого уравнения по температуре получим формулу для расчета энтальпии сольватации, которую называют уравнением Борна - Бьеррума:
Однако использование уравнения Борна – самое грубое описание процесса растворения иона. При термодинамическом рассмотрении процесса перехода ионов одного знака из газовой в жидкую фазу приходится считаться с явлениями, связанными с взаимной компенсацией эффектов для катионов и анионов. Эти эффекты могут быть одинаковыми по абсолютной величине, но разными по знаку (Ланге, Мищенко, 1930 г.). Усовершенствования расчетов, связанных с использованием уравнения Борна можно свести к следующему. 1. Считать радиус иона ri гипотетическим радиусом иона в растворе. Одно время радиусы иона в растворе пытались вычислять именно по тепловым эффектам растворения. Попытки успехом не увенчались. Пока. 2. Вводить дополнительные части в уравнение Борна. Например, по модели Латимера это добавочный член для ионов s-элементов, равный энергии растворения соответствующего инертного газа. 3. Предполагается, что вода вблизи иона сильно сжатее под действием электростатического поля иона (Бриджмент), следовательно в уравнении Борна следует использовать диэлектрическую проницаемость такой сильно сжатой воды. Метод активностей Активность и коэффициент активности являются реальными физическими величинами. Определены могут быть из эксперимента несколькими методами. Для одного и того же вещества получается хорошее совпадение данных (методом криометрии, по упругости пара, по измерению ЭДС и др.) Термодинамические уравнения для растворов электролитов, записанные через активности, подобны соответствующим уравнениям растворов неэлектролитов (температура замерзания, кипения, осмотическое давление, уравнение Рауля….). Различие заключается только в том, что 1) растворы электролитов проводят электрический ток, 2) все виды частиц раствора следует рассматривать как самостоятельные компоненты. Считается, что в сильных электролитах такими самостоятельными частицами являются катионы, анионы и молекулы растворителя. В случае слабого электролита – катионы, анионы, недиссоциированные молекулы растворенного вещества, молекулы растворителя. Для растворов электролитов вводится дополнительное условие, по которому 3) концентрации заряженных частиц в равновесных гомогенных системах связаны между собой условием электронейтральности. Условие электронейтральности:
где ni – концентрация частиц сорта i; zi – заряд частицы сорта i; е – заряд электрона. Для нейтральных частиц z = 0. Основой любой количественной термодинамической теории является уравнение функциональной зависимости энергии Гиббса с концентрацией веществ, входящих в систему. Из такого уравнения могут быть получены уравнения для описания любых других термодинамических свойств системы, т.к. энергия Гиббса является термодинамическим потенциалом. Энергия Гиббса связана с константой равновесия уравнением:
Если оставить предположение Аррениуса о характере диссоциации электролита, то константа равновесия есть константа процесса диссоциации. Для бинарного электролита, который диссоциирует по схеме
Константа равновесия с учетом закона разведения Оствальда запишется следующим образом:
Казалось бы грубая модель раствора электролита уже существует: расчет энергии Гиббса по Борну + ….еще 2 уравнения. Однако в рамках теории Аррениуса =не учитывается взаимодействие ионов с молекулами растворителя, являющееся основным в механизме диссоциации. =не рассматриваются межионные взаимодействия, включающие притяжение разноименно заряженных частиц и отталкивание одноименно заряженных. Попытки учета обоих неучтенных в модели Аррениуса взаимодействий привели к формированию современных термодинамических теорий электролитов. В основу их положен формальный подход, предложенные Льюисом в 1908 г. – через химические потенциалы. Для вещества в растворе справедливо уравнение
которое применимо для растворов электролитов. Формально уравнение химического потенциала с учетом условия электронейтральности можно применить для описания отдельно взятого иона. Однако при этом физический смысл теряется и остается только математическая функция. Следует здесь четко понимать: = невозможно в растворе выделить отдельный ион, который жил бы вне зависимости от других ионов и молекул растворителя; = невозможно внести в раствор ионы только одного знака – вместе с ними обязаны появиться противоионы и суммарная энергия Гиббса системы меняется из-за притока электронейтрального вещества s.
где s – растворенное вещество s. Связь между активностями отдельных ионов и соли (нейтрального вещества) устанавливается чисто формально из условия электронейтральности. Например для реакции
Условие электронейтральности запишется в виде
тогда
Поскольку в стандартном состоянии условие электронейтральности сохраняется, то
Тогда
т.е. активность соли в растворе формально выражается через произведение активностей ионов, на которые может формально распадаться данный электролит. Однако с помощью этой формулы определить активность каждого из ионов невозможно.
**** в любой концентрационной шкале величина активности безразмерна, размерности не имеет также и коэффициент активности, поскольку логарифм коэффициента активность есть работа переноса данного вещества из реального в идеальное состояние в энергетических единицах RT***
Концентрации ионов связаны с концентрацией соли соотношениями:
тогда
Таким образом, средняя ионная активность вещества в растворе будет определяться уравнением
То же самое, с другими обозначениями, часто встречающимися в литературе
Для описания растворов электролитов используется несимметричная система стандартных состояний, в которой за стандартное состояние растворенного вещества принимается его состояние в гипотетическом одномоляльном растворе. В зависимости от способа выражения концентрации раствора стандартное состояние выбирается таким образом, чтобы средний ионный коэффициент активности стремился к 1 при концентрации стремящейся к 0. Это требование работает при любых температуре давлении. Не следует путать гипотетический стандартный раствор с бесконечно разбавленным!!!! Для стандартного раствора
Для бесконечно разбавленного
т.е. химический потенциал = – ∞ Среднюю ионную активность и средний ионный коэффициент активности электролита определяют несколькими методами: по повышению температуры кипения раствора; по понижению температуры замерзания раствора; по давлению пара растворителя над раствором; по ЭДС гальванических элементов, по растворимости малорастворимых соединений, по электропроводности и др. Средняя ионная активности и средний коэффициент активности зависят не только от концентрации раствора, но и от заряда иона. В области низких концентраций средний ионный коэффициент активности определяется зарядом образующихся ионов и не зависит от других свойств электролитов. В разбавленных растворах средний ионный коэффициент активности зависит от следующих факторов. 1. Общая концентрация ионов в растворе и их зарядов, т.е. от ионной силы раствора I, которая рассчитывается по формуле
Правило постоянства ионной силы (Льюис, Рендал): в разбавленных растворах коэффициент активности сильного электролита одинаков для всех растворов с одной и той же ионной силой независимо от природы электролитов. Это правило справедливо при концентрациях не более 0,02М. В растворах средних и высоких концентраций правило ионной силы трансформируется, так как при более высоких значниях ионной силы усложняется характер межионного взаимодействия и проявляются индивидуальные свойства электролитов. Чем выше ионная сила раствора, тем ниже коэффициент активности. При этом в растворах с одинаковой ионной силой среднеионные коэффициенты активности в первом приближении совпадают. В предельно разбавленном растворе ионная ассоциация отсутствует, и коэффициент активности равен 1, то есть активность равна концентрации. 2. Наличие специфических взаимодействий между ионами, в первую очередь, комплексообразования. Комплексы отличаются от ионных ассоциатов тем, что ионы при образовании комплекса частично или полностью теряют гидратную оболочку и вступают в непосредственную химическую связь друг с другом. В результате комплексообразования коэффициент активности резко понижается. 3. Гидратация ионов. Наиболее сильно гидратируются ионы с малым радиусом и большим зарядом, например, Li+, Mg2+, Al3+. В концентрированных растворах их солей вся вода связана в гидратные оболочки, практически нет свободных молекул воды. Поэтому кажущаяся концентрация, или активность, выше действительной. С ростом концентрации в таких растворах наблюдается рост коэффициента активности. При этом в концентрированных растворах коэффициент активности может быть больше 1. Активность представляет собой ту эффективную концентрацию, которой должна была бы обладать реальная система, чтобы производить такие же действия, как и идеальная. Теория активности пытается сохранить обычные формулировки термодинамических соотношений при помощи обычного вида термодинамических потенциалов, вводя понятие об эффективной активности, зависящей от окружающих условий и концентраций. Однако в рамках чистой термодинамики невозможно определить вид функциональной зависимости коэффициента от концентрации. Данную зависимость установить можно двумя путями: 1. Из экспериментальных данных выводить закономерность зависимости коэффициента активности от концентрации и температуры 2. Вычислять значение коэффициента активности методами статистической механики. Теория Дебая-Хюккеля В основе теории Дебая-Хюккеля положено представление о том, что отклонение свойств реального раствора электролита от идеального связано с электростатическим взаимодействием ионов между собой. Поэтому для коэффициента активности иона в растворе можно записать
или
где разность химических потенциалов иона в случае чисто кулоновского взаимодействия выражается в виде электрической работы переноса единичного заряда ezi из идеального в реальное состояние. Для расчета коэффициента активности иона необходимо знать · разность потенциалов между двумя соседними ионами или · распределение поля вокруг одного, заранее выбранного в качестве центрального, иона. Решение данной задачи Дебай и Хюккель выполнили при следующих допущениях 1. Электролит в растворе диссоциирован полностью 2. Концентрацию ионов рассчитывают по аналитической концентрации электролита 3. Растворитель считать непрерывной средой с определенной диэлектрической проницаемостью 4. Диэлектрическая проницаемость растворителя не зависит от концентрации раствора. 5. На диэлектрическую проницаемость растворителя не влияет электростатическое поле иона 6. Диэлектрическая проницаемость раствора равна таковой для расворителя 7. Ион в растворе – точечный заряд. 8. Распределение ионов в растворе определяется тепловой энергией и энергией электростатического взаимодействия, т.е. подчиняется классической статистике Больцмана:
где n0 – концентрация ионов в единице объема раствора; ni – концентрация ионов в объеме шарового слоя, проведенного вокруг центра тяжести иона, выбранного в качестве центрального; φ – среднее значение потенциала шарового слоя (ионная атмосфера). 9. Из всех видов межионного взаимодействия учитывать только электростатическую составляющую. 10. Каждый ион в растворе окружен сферой из противоположно заряженных ионов, суммарный заряд которых уравновешивает заряд центрального иона. Остается неучтенным: сольватация, поляризуемость, особенности строения ионов и т.д.
Уравнение связи между потенциалом φ в данной точке поля и зарядом q, создающим это поле, основано на распределении Гаусса через уравнение Пуассона (
где r – расстояние от центра она до рассматриваемой точки; ε0, ε – диэлектрические проницаемости вакуума и рассматриваемой среды; ρ – объемная плотность заряда. Объемная плотность заряда, с учетом распределения Больцмана, будет равна
Основное уравнение теории Дебая-Хюккеля в дифференциальной форме
После разложения в ряд и учета только первых двух членов ряда получится упрощенная версия основного уравнения Д-Х
Величина χ имеет размерность обратной длины. Эту величину называют Дебаевским радиусом или радиусом ионной атмосферы. Представляет собой характерное расстояние, на котором полностью экранируется поле данного иона. После интегрирования и преобразований получается интегральная форма основного уравнения Дебая-Хюккеля
Данное уравнение используется для определения усредненного потенциала, создаваемого ионом в точке, отстоящей на расстоянии r от иона в отсутствии внешних сил. а – расстояние наибольшего сближения. Для вычисления работы электростатического взаимодействия ионов необходимо учесть вклады в электростатическое поле как от ионной атмосферы, так и от самого центрального иона, вокруг которого сформировалась ионная сфера. Потенциал в точке, расположенной на расстоянии r от центра изолированного иона, находящегося в среде с диэлектрической проницаемостью ε∙ε0, имеет вид
Тогда разность потенциалов при r>a
Если r = a, то уравнение имеет гораздо более простой вид
Данное выражение представляет собой изменение потенциала иона при внесении в его поле соседних ионов или электрическую работу, которая определяет коэффициент активности в модели Дебая-Хюккеля Уравнение коэффициента активности иона в растворах электролитов:
И в более компактной форме
где
Поскольку коэффициент активности отдельного иона нельзя измерить экспериментально, можно использовать понятие среднего ионного коэффициента активности:
Если слагаемое

Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2024 stydopedia.ru Все материалы защищены законодательством РФ.
|
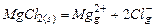




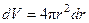 полная энергия поля равна
полная энергия поля равна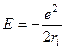





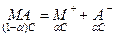

 ,
, ,
,


 .
.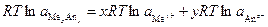


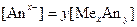







 ,
,
 ):
):
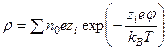





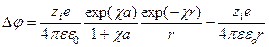



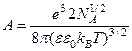 и
и 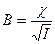

 , то уравнение для среднего ионного коэффициента активности принимает более простой вид
, то уравнение для среднего ионного коэффициента активности принимает более простой вид