
|
|
Теория Тафеля (замедленная рекомбинация)В 1905 г. Тафель предположил, что процесс катодного восстановления водорода протекает по схеме: 1) Н3О+ + e ↔ Надс+ Н2О; реакция Фольмера 2) Надс+ Надс ¾® H2, реакция Тафеля а замедленной является стадия рекомбинации. Выведем, согласно этой теории, зависимость величины перенапряжения разряда ионов водорода от плотности катодного тока. В отсутствие внешнего тока φ = φ0 + при его пропускании φi= φ0 + Тогда: hН = φ – φi = i2 = k2 i–2 = k–2 где i2 и i–2 - скорость прямой и обратной реакции стадии рекомбинации. Очевидно, что i2 = k2 ik = k2 hН = При достаточно высоких плотностях катодного тока ik >>i0 и hН = hН = a + 2,3 Из уравнения (76) получаем 3 критериальных величины, характерных для механизма Фольмера-Тафеля с замедленной реакцией Тафеля: (дh /dlgi)Ci = bk = 2,3RT/(2F) = 0,029 В =29 мВ (18оС) (дh /dlg (dlgi/dlg Еще 3 критериальные величины для этого механизма получим из выражения для φi. η = φравн – φi, откуда φi = φравн – η С учетом φравн = (2,3RT/F)lg CH+ (при РН2 = const) и выражения для hН имеем: φi = (2,3RT/F)lg CH+ - а - 2,3 (дφ /dlgi)Ci = bk= -2,3 (дφ /dlg (dlgi/dlg
Теория Фольмера
Н3О+ + e ¾® Надс+ Н2О; кислая среда Надс+ Надс ↔ H2,
Н2О + e ¾® ОН– + Надс; щелочная и нейтральная среды Надс+ Надс ↔ H2,
Для вывода кинетического уравнения необходимо учесть, что в случае разряда протона энергия активации процесса зависит от потенциала электрода: Ек = Е0 + aφF Соответственно для обратной реакции имеем: Еа = Е0 - bφF, где a и b - числа переноса, связанные, как указывалось ранее, зависимостью a + b = 1. Для реакции разряда:
ik = k CH+
ln ik = lnk + lnCH+ – φ = const + 2,3 Из последнего выражения получаем 3 критериальные величины, характерные для этого механизма: (дφ /dlgi)Ci = bk= -2,3 (дφ /dlg (dlgi/dlg
Найдем связь перенапряжения реакции разряда ионов водорода с плотностью катодного тока. η = φравн – φi, η = (2,3RT/F)lg CH+ - const - 2,3 или h = const – 2,3 Из последнего выражения находим еще 3 критериальные величины, характерные для данного механизма:
(дh /dlgi)Ci = bk = 2,3RT/(αF) = 0,116 В =116 мВ (18оС) (дh /dlg (dlgi/dlg
При рассмотрении этой реакции А.Н.Фрумкин учел влияние строения двойного электрического слоя на кинетику процесса, за которую ответственен только потенциал плотной ее части. Если падение потенциала в диффузной части двойного слоя обозначить за Y1 то
i2 = k1 Если поверхность электрода заряжена отрицательно, то есть его потенциал отрицательнее потенциала нулевого заряда, то Y1< 0 и концентрация катионов в двойном слое больше, чем в объеме:
ik = k CH+
ik = k CH+ Откуда после логарифмирования получаем: φ = const + 2,3 Получаем 3 критериальные величины: (дφ/дlgCH+ ) Ci,i = (дφ/дlgik) Ci = - (дlgik/дlgCH+)φ,Ci =1,0.
Найдем зависимость перенапряжения разряда ионов водорода от величины ik: h = φр – φ, φр = 2,3 h = 2,3 h = const’ – 2,3 При a=0,5 и Т = 291К (дh/дlgCH+ )Ci,i = (дh/дlgik)Ci = (дlgik/дlgCH+)h,Ci = 1–a = 0,5
Независимость h от CH+ в растворах кислот обусловлена взаимной компенсацией 2-го и 3-го членов последнего уравнения. Когда ионная сила раствора постоянна, то y1 – const. Подобная компенсация отсутствует, и величина (дh/дlgCH+) = –58 мВ. Это вытекает из следующего. При не очень высокой концентрации электролита с одновалентным катионом и при значениях электродного потенциала, более отрицательных, чем потенциал нулевого заряда (как это обычно имеет место при катодном выделении водорода), величина ψ1 подчиняется следующему уравнению:
где С - общая концентрация одно-одновалентного электролита в растворе, В - постоянная. Подстановка (78) в (77) приводит к уравнению:
где a =const’ - В . Представляют интерес три частных случая: 1. Разбавленные растворы кислоты без добавки соли: С =СH+, тогда (79) принимает вид:
т.е. перенапряжение не зависит от концентрации кислоты (подтверждается данными для НС1, когда концентрация не превышает 0,1 моль/л). Объясняется это компенсацией двух эффектов: с ростом концентрации кислоты увеличивается концентрация реагирующего вещества, что должно уменьшать η, и одновременно с увеличением концентрации происходит сжатие двойного слоя (сдвиг ψ1в положительную сторону) и увеличение перенапряжения. 2. Общая концентрация раствора остается постоянной СHCl +CKCl = const, но изменяется отношение CHCl / СKCl. При этом
где const1= или при α =0,5; т.е. перенапряжение должно возрастать на 3. Концентрация кислоты постоянна, а концентрация соли изменяется. При α = 0,5 из (79) получаем
где const2= т.е. перенапряжение должно возрастать на
Возможен и иной путь удаления атомарного водорода, кроме рекомбинации: Надс + Н3О+ + е ¾®Н2О + Н2 (реакция Гейровского). Тогда процесс выделения водорода протекает следующим образом: 1) Н3О+ + e ↔ Надс+ Н2О 2) Надс + Н3О+ + е ¾®Н2О + Н2 Для 2-ой стадии: i2 = k2 CH+ θ где Q пропорционально CHадс. Для первой квазиравновесной стадии справедливо уравнение Нернста: φ = φо + (RT/F)lg(CH+/θ), откуда CH+/θ = exp( θ = k1 CH+ exp( Подставив θ в i2, получим i2 = k и окончательно ik = k ln ik = ln k + 2ln CH+ - (1+α)φF/(RT) или lg ik = lg k + 2lg CH+ - (1+α)φF/(2,3RT), откуда φ = Из последнего выражения получаем 3 критериальные величины, характерные для этого механизма: (дφ/дlgCH+ ) Ci,i = (дφ/дlgik) Ci = - (дlgik/дlgCH+)φ,Ci =2,0.
Найдем связь перенапряжения реакции разряда ионов водорода с плотностью катодного тока. η = φравн – φi, η = (2,3RT/F)lg CH+ - const - или η = const’ - Из последнего выражения находим еще 3 критериальные величины, характерные для данного механизма при α=0,5 и Т=291К: (дh/дlgCH+ )Ci,i = - (дh/дlgik)Ci = (дlgik/дlgCH+)h,Ci = 1–a = 0,5 Таким образом, рассмотренные 3 механизма реакции катодного выделения водорода вполне различимы по своим критериальным величинам. На металлах с высоким hнзамедленным является разряд (Hg, Zn, Cd). На металлах с низким hнпо мнению некоторых авторов (Л.И.Антропов), замедленной является рекомбинация, но bк = 100 мВ (по Л.И.Антропову – 60 мВ). На Ni, по А.Н.Фрумкину и П.Д.Луковцеву, имеются два рода участков. На одних замедлен разряд, на других - рекомбинация. По Л.И.Антропову, замедлена только рекомбинация. На Fe, по Бокрису, в кислых средах замедлен разряд, в щелочных при низких hн- рекомбинация, при высоких hн - разряд, по И.А.Багоцкой, в щелочных средах - замедленный разряд на неоднородной поверхности, по В. Пасту и З.А.Иофа, скорости разряда и рекомбинации соизмеримы. На олове в щелочных средах, по В.Пасту, замедлен разряд. Относительно влияния сольватной формы протона (H3O+,ROH2+, CH3CNH+,(CH3)2SOH+ и др.) на величину перенапряжения водорода существуют различные точки зрения. Л.И.Антропов указывает, что hн является функцией свободной энергии сольватации и, следовательно, природа сольватирующего агента (растворителя) влияет на hн. Б.Б.Дамаскин, О.А.Петрий и Л.И.Кришталик отрицают влияние DGsolv на hн. Авторами настоящего пособия был проведен ряд подобных исследований в этанольных и этиленгликолевых растворах НС1 на железе армко. Этанольные растворы НС1 При введении воды в 1 М растворы НС1 (рис. 34) происходит систематическое понижение тафелевского коэффициента наклона, который достигает минимального значения 28 - 30 мВ в присутствии 2 мас. %Н2О.
Рис 34. Зависимость концентрации ионов Н+ в форме ионов гидроксония (1), величины коэффициента «bk» уравнения Тафеля (2а) и перенапряжения водорода (2б) в 1 М этанольных (а) и этиленгликолевых (б) растворах НС1 от содержания воды в смешанном растворителе при 200 С. Дальнейшее повышение Отметим, что в водных солянокислых средах на железе, по нашим данным, замедленной является стадия разряда, так как при постоянной ионной силе наблюдаются следующие кинетические параметры: (дjН /дlgi)Ci = 100 мВ, (дlgi/дpH) (дlgi/дpH) Зависимость перенапряжения водорода на железе от содержания воды в этиленгликолевых растворах показана на рис. 34 (bk –90–100 мВ). Легко видеть, что экстремальную зависимость hн = f(CH2O) нельзя объяснить за счет существования различных сольватных форм разряжающегося протона (С2Н4(ОН)2Н+ и Н3О+). В условно безводных этиленгликолевых растворах перенапряжение водорода понижается с ростом CH+. Кинетические параметры процесса равны: (дhн/дlgi)Ci = 100 мВ, (дlgi/дlgCH+) (дlgi/дlgCH+)j,Сi = 0,9, (дhн/дlgCH+) Ci= – 67мВ, и находятся в согласии с теорией замедленного разряда. В присутствии 2 – 10 % Н2О картина меняется. hнне зависит от CH+ как при поддержании постоянной ионной силы, так и в отсутствие солевой добавки. Величина (дhн/дlgi)Ci остается близкой к 100 мВ. Видимо, в этих средах замедленной является рекомбинация. Изменение характера замедленной стадии не связано с сольватной формой разряжающегося протона, так как разряд Н3О+ наблюдается в присутствии 10 % Н2О и в чисто водных растворах. Следовательно, в этиленгликоле и его водных смесях (как и в С2Н5ОН) hни природа замедленной стадии не зависят от сольватной формы разряжающегося протона и его реальной энергии сольватации. Исследования проведенные в последние годы под руководством авторов на кафедре аналитической и неорганической химии Тамбовского государственного университета им. Г.Р. Державина (ж. Электрохимия 2001-2009 гг, J. Electroanal. Chem. 2004-2009 гг) показали, что изменение состава смешанного растворителя позволяет целенаправленно менять природу лимитирующей стадии реакции выделения водорода (РВВ). В связи с этим, в соответствии с требованиями электрохимической кинетики меняются и кинетические параметры суммарного процесса. Часто определяющую роль в подобных эффектах играет природа растворителя, адсорбирующегося на металлической поверхности. Такая картина наблюдается, в частности. Для РВВ на железе в системах С2Н4(ОН)2 – Н2О-НС1 и С2Н5ОН - Н2О-НС1 с постоянной ионной силой. Кроме того, кинетические параметры РВВ зависят от энергетического состояния поверхности металла и степени заполнения ее активных центров адсорбированным водородом. Для железа кинетические параметры реакции выделения водорода обобщены в таблице 3 (опубликованной впервые в подобном виде в ж. Электрохимия, 2001). Для других металлов подобные обобщения отсутствуют.
Таблица 4. Теоретические кинетические параметры катодного восстановления ионов водорода, соответствующие различным условиям протекания процесса (Ψ1 = 0, α = β = 0,5, Т = 20˚С).
*[i] – А/м2, [С] – М; **параметр является функцией потенциала электрода; ***числитель – активированная адсорбция, знаменатель – неактивированная. КОНЦЕНТРАЦИОННАЯ ПОЛЯРИЗАЦИЯ До сих пор мы рассматривали процессы, протекающие в кинетической области, когда скорость реакции определяется собственно скоростью переноса заряда. Возникающая в этих условиях поляризация получила название электрохимической. Однако имеется значительное количество процессов, характеризующихся большим током обмена и, следовательно, протекающих с очень малым перенапряжением переноса, то есть перенапряжением, обусловленным переносом заряда через границу раздела фаз металл-раствор. В этом случае при протекании анодного растворения металла в приэлектродном слое появляется значительное количество ионов - продуктов растворения. Возникают условия, при которых концентрация ионов в приэлектродном слое зависит от скорости отвода (или подвода) их в объем раствора. В первый момент она возрастает во времени, а затем становится постоянной, то есть достигаются условия стационарной диффузии. При наличии в растворе фонового (индифферентного) электролита, определяющего электропроводность раствора, вкладом в перенос тока ионами (изменение концентрации которых мы рассматриваем) под действием электрического поля (миграция) можно пренебречь. В этом случае оценивают их диффузионный перенос. Определенная им плотность тока i (сила тока, отнесенная к единице поверхности) равна: i = nFD где п - заряд иона, F - число Фарадея, D - коэффициент диффузии, Cs - концентрация ионов в приэлектродном слое, С0 - концентрация ионов в объеме раствора, d - толщина диффузионного слоя, на протяжении которого концентрация диффундирующих ионов меняется от Cs до С0. В случае процессов коррозии и анодной ионизации С0 обычно равна нулю. Тогда уравнение для плотности тока диффузии принимает вид: iдиф = nFD В отсутствие перенапряжения переноса потенциал электрода определяется только концентрацией одноименных ионов, близкой к равновесной. В этом случае он выражается уравнением Нернста: φ= φ0 + Совершенно очевидно, что поверхностная концентрация диффундирующих ионов зависит от плотности анодного тока, возрастая с ее величиной. В общем случае: СS = Подставив выражение (83) в уравнение Нернста, можно получить связь между ia и СS, то есть ia = F(CS), так как анодный ток целиком определяется током диффузии: φ = φ0 + φ = φ0 + φ0 + φ = const + Не трудно видеть, что в этом случае поляризационная кривая в координатах φ– lgi также выражается прямой линией. Однако ее наклон не связан с коэффициентом переноса. Он равен 0,058/n, и по величине (дφ/дlgi)Ci можно определить величину заряда переходящих в раствор ионов. В ряде случаев переход ионов в раствор сопровождается последующим комплексообразованием или комплексные частицы образуются непосредственно в акте ионизации металла. Можно полагать, что в таких условиях скорость анодного процесса растворения металла будет связана с величиной константы стойкости соответствующих комплексных ионов. В самом деле, пусть некоторый металлический электрод обратим относительно собственных ионов. Процесс анодного растворения протекает по уравнению Ме « Меn+ + пе. Комплексообразование происходит непосредственно в двойном электрическом слое и протекает по уравнению: Меn+ + kАm–«МеА Металл же, естественно, обратим только относительно простых ионов. Картина может быть и принципиально иной. Пусть на поверхности металлического электрода образуется поверхностный промежуточный комплекс типа МеА МеА Возникающие комплексные частицы должны находиться в растворе в равновесии с простыми типа Меn+ в соответствии с константой равновесия (Кс).Таким образом, оба эти подхода приводят к одной и той же физической картине. Далее будем считать, что в изотермических условиях в двойном электрическом слое и в объеме раствора величина Кс неодинакова. Тогда потенциал металлического электрода, обратимый относительно ионов Меn+, не зависит от того, по какому механизму образуются простые ионы и за счет чего достигается их равновесная концентрация - происходит ли образование комплексных ионов после перехода простых в раствор или за счет разрушения первичных комплексов. Константа равновесия (далее константа стойкости Кст) процесса (84) имеет вид: Кст =
Очевидно, что суммарная активность ионов, образующихся на основе растворяющегося металла, равна:
Обозначим ее через В. Тогда
В =
Равновесный потенциал электрода может быть выражен уравнением φ = φ0 + Постулируем активность ионов растворяющегося металла в объеме раствора
Анодный ток, определяемый диффузионным отводом ионов
С другой стороны, ионы Меn+ отводятся посредством комплексообразования. Формально примем величину этого процесса в токовых единицах равной:
Тогда общая скорость отвода от поверхности электрода простых ионов в электрических величинах равна:
С учетом выполнимости уравнения Нерста можно записать:
Очевидно, что при значительной величине Кст: a2>>a1 и
Выразим В из (88) и подставим в (87). Это дает
Решая уравнение (89) относительно φа, получим: φа = const – При величине и, равной 1, имеем φа = const – Следовательно, с ростом Кстпри φа = const в условиях диффузионного контроля процесса скорость ионизации металла должна возрастать. В самом деле, из уравнения (90) следует, что при постоянстве φа lniа является линейной функцией ln а величина kхарактеризует наклон прямой. И.К.Маршаков с сотрудниками проверили выполнимость последней зависимости при ионизации меди в растворах состава: С М НС1 + Х М КС1 при рН = 2. Они получили линейную зависимость с k=3 и пришли к выводу, что этом случае образуются комплексные ионы CuCl32– ( Проверки уравнения (90) по Kст в научной литературе нам обнаружить не удалось. Если при равновесном потенциале в отсутствие внешнего тока концентрация потенциалопределяющих ионов Со, а при его наложении CS,то можно записать: φ = φ0 + φ = φ0 + Разность Еi>0 – Еi=0 представляет собой концентрационную поляризацию hk. hkонц = hkонц = Величина диффузионного тока равна:
Его предельная величина наблюдается при С0 ¾® 0
Отсюда: Подставив последнее выражение в уравнение (91), получим: hконц = В случае катодного процесса
hkонц =
Выражение для hконцостается прежним (уравнение (92)): hконц = Концентрационная поляризация анодного процесса способствует сдвигу потенциала электрода в положительную сторону, катодного – вотрицательную. В общем случае возможны чисто диффузионные и чисто кинетические процессы. В первом случае их скорость контролируется транспортом участвующего в процессе вещества от электрода или к электроду, во втором - она определяется природой протекающей реакции. В первом случае возникает перенапряжение диффузии hд во втором - перенапряжение перехода hп. Возможны и промежуточные случаи, когда hд и hп соизмеримы. В этом случае, чтобы найти hп нужно из общего сдвига потенциала вычесть hд, рассчитанное по уравнению (92), что используется при исследовании катодных реакций. Наличию диффузионного контроля способствуют большие токи обмена, высокие плотности внешнего тока, малая величина коэффициента диффузии, малая концентрация окислителя при катодных реакциях. Скорость процессов, контролируемых диффузией, существенно зависит от гидродинамических условий в диффузионном слое. В случае процессов, протекающих на вращающемся дисковом электроде, сила тока выражается зависимостью: i = 0,62nFD2/3w1/2v–1/6C, где D - коэффициент диффузии, w - скорость вращения диска, v - кинематическая вязкость, С - концентрация диффундирующих частиц. При постоянстве всех сомножителей, кроме w, уравнение принимает очень простой вид: i = k В координатах i – В общем случае экспериментально наблюдаемая зависимость i=f(w1/2) имеет вид, показанный на рис 35б. На кривой можно выделить 3 участка: аб - область чисто диффузионного контроля. Кинетика процесса определяется только транспортными ограничениями. 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2024 stydopedia.ru Все материалы защищены законодательством РФ.
|



 ;
;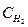 ;
; – k–2
– k–2  ;
;  ;
; =
=  .
. ; hН = 2,3
; hН = 2,3  – 2,3
– 2,3  ;
;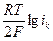 (76).
(76). = 0 ;
= 0 ; = nH+= 0
= nH+= 0 = -0,029 В = - 29 мВ;
= -0,029 В = - 29 мВ; = nH+ = 2.
= nH+ = 2. В 1930 г. Эрдей-Груз и Фольмер предположили, что при катодном восстановлении протонов замедленной является стадия разряда, а удаление атомарного водорода происходит посредством рекомбинации по схемам:
В 1930 г. Эрдей-Груз и Фольмер предположили, что при катодном восстановлении протонов замедленной является стадия разряда, а удаление атомарного водорода происходит посредством рекомбинации по схемам:
 , откуда
, откуда ;
; lgCH+ – 2,3
lgCH+ – 2,3  lgCH+ + 2,3
lgCH+ + 2,3 
 и
и  .
.
 ;
;
 – 2,3
– 2,3 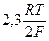 = 116 мВ;
= 116 мВ; = –116 мВ;
= –116 мВ;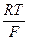 lgCH+ (при рН2 =1 ат)
lgCH+ (при рН2 =1 ат) lgCH+ +
lgCH+ +  (78)
(78) (79)
(79) (80)
(80) (81)
(81) ,
,
 = 58 мВ (при 18° С) при увеличении рН на единицу.
= 58 мВ (при 18° С) при увеличении рН на единицу. (82)
(82) ,
, ;
; ) = k1’ exp(
) = k1’ exp(  )
) )
) exp(
exp(  )
) lgk +
lgk + 
 lg CH+ +
lg CH+ + 
 до 3 и более процентов вновь увеличивает bк до 100 - 105 мВ (т.е. до исходного значения в условно безводном спирте). Это затрудняет сопоставление величин hн в смешанных растворителях разного состава, так как Dhн является функцией плотности тока. Однако можно показать, что изменение bк не связано с изменением формы разряжающегося протона. Согласно экспериментальным данным (рис. 34), значение (дh/дlgi)Ci в растворах с единственной формой сольватированного протона (С2Н5ОН2+ или Н3О+) характеризуется одинаковой величиной. Минимум bк наблюдается, когда эти формы существуют в соизмеримых количествах. В этанольных растворах НС1 + LiCl с постоянной ионной силой величина (дlgi/дlgCH+)h,Ci равна нулю независимо от
до 3 и более процентов вновь увеличивает bк до 100 - 105 мВ (т.е. до исходного значения в условно безводном спирте). Это затрудняет сопоставление величин hн в смешанных растворителях разного состава, так как Dhн является функцией плотности тока. Однако можно показать, что изменение bк не связано с изменением формы разряжающегося протона. Согласно экспериментальным данным (рис. 34), значение (дh/дlgi)Ci в растворах с единственной формой сольватированного протона (С2Н5ОН2+ или Н3О+) характеризуется одинаковой величиной. Минимум bк наблюдается, когда эти формы существуют в соизмеримых количествах. В этанольных растворах НС1 + LiCl с постоянной ионной силой величина (дlgi/дlgCH+)h,Ci равна нулю независимо от  ,Ci = –0,55,
,Ci = –0,55, В
В
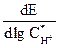 В
В

 В
В
 B
B

 ,
, ,
,
 (83)
(83) ;
; +
+ 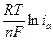 ;
; . (84)
. (84) . В процессе анодной ионизации этот комплекс переходит в раствор в виде комплексных ионов по уравнению:
. В процессе анодной ионизации этот комплекс переходит в раствор в виде комплексных ионов по уравнению: ;
; .
.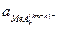 +
+  .
. +
+  ;
; .
. .
. »0. Тогда в условиях диффузионного контроля отвода продуктов ионизации, как указывалось ранее, имеем:
»0. Тогда в условиях диффузионного контроля отвода продуктов ионизации, как указывалось ранее, имеем: =
=  .
. =
=  .
. =
=  .
. ;
;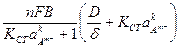 . (86)
. (86) . (87)
. (87)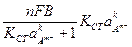 . (88)
. (88)
 .(89)
.(89)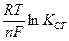 –
–  +
+  –
–  +
+  . (90)
. (90) , так как
, так как ;
; . (91)
. (91) =
=  .
.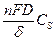 ,
, =
=  =
= 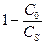 .
. = 1 –
= 1 – 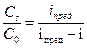
 . (92)
. (92) =
=  , так как
, так как  ;
; , когда СS ® 0;
, когда СS ® 0; .
. =
=  ,
,  =
=  –
– 
 .
.