|
|
Получение эфирного масла из тминаОсновными способами получения эфирных масел для пищевых целей являются перегонка с водяным паром (или водой) и холодное прессование, а также экстракция. Эффективным способом непрерывного извлечения эфирных масел в лабораторных условиях является сочетание паровой дистилляции и экстракции растворителем. В качестве экстрагента чаще всего используют этиловый спирт и очищенный петролейный эфир. Другие растворители - хлороформ, диэтиловый эфир сравнительно дороги, кроме того, их применение приводит к получению сильно окрашенных продуктов. При выборе растворителя следует учитывать его чистоту, летучесть и “нейтральность”. В растворителе не должны присутствовать токсичные вещества, а также вещества, обладающие запахом и изменяющие аромат эфирного масла. Летучесть растворителя определяет температуру, при которой происходит экстракция. Чем ниже температура кипения растворителя, тем в более мягких условиях происходит процесс извлечения эфирного масла и дальнейшего удаления растворителя. “Нейтральность” растворителя не позволяет проходить химическим реакциям в процессе выделения эфирного масла. Недостатком этилового спирта является способность этерифицировать содержащиеся в растительных тканях органические и жирные кислоты, при этом образуются весьма пахучие этиловые эфиры, способные сильно изменить аромат эфирного масла. Часто применяющийся петролейный эфир удовлетворяет большинству требований к экстракции. Сравнительно новым способом извлечения эфирных масел некоторых цветковых растений является динамическая адсорбция, то есть поглощение ароматических веществ активированным углемили другими твердыми адсорбентами. Для этого лепестки цветков загружают в камеру и продувают их увлажненным воздухом. Насыщенный ароматами воздух направляется в адсорбер с активированным углем, где происходит насыщение угля эфирным маслом. Затем уголь промывают диэтиловым эфиром и эфир выпаривают.
Оборудование: электрический сушильный шкаф весы лабораторные общего назначения колбы холодильник аппарат Сокслета термометры лабораторные на интервал температур 50 – 200 °С цилиндры Ход работы: Способ 1. 20 г высушенной зелени тмина подвергают перегонке с водяным паром по способу, описанному для мальтола. Отгонку ведут в течение 1,5-2 часов. На поверхности дистиллята образуется эфирный слой, который тщательно отделяют от водной фазы и промывают водой. Весовым методом определяют выход эфирного масла из расчета на исходное сырье. Способ 2. 20 г высушенной зелени тмина экстрагируют 100 мл гексана в аппарате Сокслета в течение 2-3 часов. По завершении экстрагирования гексан отгоняют под пониженным давлением. Определяют выход эфирных масел и сравнивают масла, полученные двумя способами (выход, органолептические свойства). Контрольные вопросы: 1. Каким требованиям должен отвечать растворитель, используемый в качестве экстрагента при получении эфирных масел? 2. Дайте сравнительную характеристику разных способов получения эфирных масел. 3. Опишите метод динамической адсорбции.
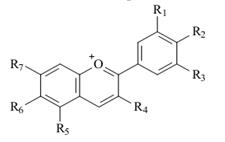
Цвет Молекулы органических соединений могут иметь σ-, π- и n-электроны. Простую, одинарную связь образуют σ-электроны. Они прочно удерживаются атомом, и для их возбуждения необходима большая энергия. Например, в молекулах насыщенных углеводородов связи σ-С–Н и σ-С–С переходят в возбужденное состояние от порций энергии более 800 кДж/моль, что соответствует энергии фотонов дальней ультрафиолетовой части спектра, поэтому молекулы насыщенных углеводородов бесцветны. В ненасыщенных углеводородах двойная связь образуется π-электронами. π–Электроны более подвижны, легче возбуждаются, и их переход на высший энергетический уровень требует меньших затрат энергии. Однако углеводороды с одной двойной связью или с несколькими изолированными (разобщенными) двойными связями бесцветны, т. к. для их возбуждения также необходима энергия фотонов ультрафиолетовой части спектра. Сопряжение двойных связей повышает подвижность π-электронов. В органическом соединении с длинной цепочкой сопряженных двойных связей π-электроны образуют единое электронное облако, принадлежащее уже не отдельным атомам, а всей молекуле в целом. Чем длиннее цепочка сопряженных двойных связей, тем подвижнее π-электроны и тем меньшие порции энергии нужны для их возбуждения и перевода на высший энергетический уровень. Полосы поглощения, появление которых вызывается электронными (π→π*) -переходами, обусловленными наличием сопряженной системы в целом, получили название K-полос (от слова «конъюгация» - сопряжение). Отличительной особенностью K-полос является высокая интенсивность (εмакс = 10 000÷200 000). В длинной цепи сопряжения энергия возбуждения снижается до значений, соответствующих энергии фотонов видимой части спектра (K-полоса смещается в длинноволновую часть спектра), и соединение воспринимается человеческим глазом как окрашенное. Например, в молекуле ликопина — красящего вещества томатов — содержится цепочка из одиннадцати сопряженных двойных связей, и длина волны поглощаемого света (λмакс = 506 нм) обусловливает ярко-красный цвет с синеватым оттенком. Особое значение для химии красящих веществ имеют замкнутые системы сопряженных двойных связей - ароматические ядра. Увеличение числа π-электронов в молекулах ароматических соединений оказывает на цвет такое же влияние, как удлинение сопряженной цепочки в алифатических соединениях: энергия возбуждения снижается и поглощение смещается в длинноволновую область. На электронный спектр соединения влияние оказывает также замена атомов углерода на гетероатомы в молекулах с сопряженными двойными связями. Так как гетероатомы (азот, кислород, сера и др.) имеют неподеленные электронные пары, в молекулах возникает возможность иных (помимо σ→σ* и π→π*) электронных переходов. Неподеленные электроны в основном состоянии молекулы занимают несвязывающие n-орбитали, энергетический уровень которых является для простых молекул промежуточным между уровнями связывающих и разрыхляющих орбиталей. Вследствие этого переход одного электрона с несвязывающей n-орбитали на разрыхляющую орбиталь требует значительно меньших затрат энергии, чем соответствующий переход со связывающих σ- и π-орбиталей. Наиболее длинноволновая полоса поглощения, возникающая в результате (n→π*)-перехода, называется R-полосой (от слова «радикал»). Отличительной особенностью R-полос является низкая интенсивность: обычно εмакс ≤ 100 и не бывает больше 2000. Причиной этого является различная симметрия орбиталей π-электронов гетероатома и π-электронов связи: оси симметрии этих орбиталей взаимно перпендикулярны, и нет условий для взаимного перекрывания орбиталей. В таких случаях вероятность электронного перехода мала (запрет по симметрии), а, следовательно, мала и интенсивность полосы поглощения. В отличие от гетероароматических соединений, поглощение которых сравнительно мало отличается от поглощения соответствующих ароматических соединений, замена атомов углерода гетероатомами в алифатических участках сопряженных систем часто весьма сильно сказывается на положении полосы поглощения. Однако батохромное смещение длинноволновой полосы поглощения происходит лишь в том случае, если гетероатом, замещающий данный атом углерода, более электроотрицателен по сравнению с углеродом. Если электроотрицательность гетероатома меньше, чем у углерода, полоса поглощения смещается в коротковолновую часть спектра. Переход молекулы с сопряженными двойными связями в возбужденное состояние связан с ее поляризацией, т. е. с появлением положительного заряда на одном конце молекулы и отрицательного - на другом. Введение в такую молекулу заместителей вызывает постоянное, не зависящее от поглощения света, смещение π-электронов. Заместители делятся на две группы: электронодонорные (ЭД) и электроноакцепторные (ЭА). Электронодонорные заместители обладают неподеленными электронами (π-электронами), способными вступать во взаимодействие с π-электронами цепочки сопряженных двойных связей, отталкивая их и, в конечном счете, включаясь в общую π-электронную систему молекулы. В молекулах ароматических соединений, содержащих сильные ЭД-заместители, возможны электронные переходы, сопровождающиеся переносом неподеленных электронов заместителя на ароматическое кольцо; соответствующие полосы поглощения называются полосами переноса заряда (ПЗ). Полосы ПЗ часто сливаются с полосами (π→π*)-переходов в ароматическом кольце, которые под влиянием заместителей претерпевают батохромный сдвиг по сравнению с незамещенным соединением. Наиболее важными ЭД-заместителями являются свободные и замещенные амино-, гидрокси- и меркаптогруппы. ЭА-Заместителями называют такие заместители, которые способны притягивать к себе электроны. Они содержат двойную связь, и их влияние на углубление окраски органических соединений особенно сильно сказывается в том случае, если они связаны с сопряженной системой двойных связей. К этим заместителям относятся: нитрогруппа (−NO2), нитрозогруппа (−N=O), карбонильная группа [−C(O)−], кетониминная (хинониминная) группа [−C(О)NH−] и др. Подобно ЭД-заместителям, они создают некоторое постоянное, не зависящее от действия света смещение π-электронов в сопряженной системе, усиливают их делокализацию в основном состоянии молекулы и увеличивают вклад полярной структуры. Это приводит к уменьшению энергии возбуждения и соответственно к сдвигу полосы поглощения в длинноволновую область. При введении сильных ЭА-заместителей в ароматическое кольцо возникает возможность электронных переходов с переносом π-электронов ароматического кольца на ЭА-группу, и в электронном спектре появляются соответствующие полосы ПЗ. Особенно сильное влияние на электронный спектр оказывают ЭД- и ЭА-заместители при одновременном действии, если они находятся на противоположных концах цепи сопряжения. Совместное действие поляризующих заместителей противоположного характера приводит обычно к резкому сдвигу полосы поглощения в сторону более длинных волн не только по сравнению с незамещенным соединением, но и с любым из соответствующих монозамещенных. Делая π-электронную систему более подвижной, ЭД- и ЭА-заместители увеличивают вероятность электронных переходов, т. е. вероятность избирательного поглощения фотонов, переводящих молекулу из основного состояния в возбужденное. Это приводит к увеличению интенсивности поглощения света, т. е. увеличивается интенсивность окраски соединения. Высокая интенсивность поглощения (высокий мольный коэффициент поглощения, εмакс) является типичным признаком красителей, отличающим их от других соединений, обладающих окраской. Влияние поляризующих ЭД- и ЭА-заместителей в молекулах органических соединений может быть усилено или ослаблено ионизацией. Для красителей наибольшее значение имеет ионизация таких ЭД-заместителей, как гидрокcигруппа (–ОН), аминогруппа (–NН2), меркаптогруппа (–SН), и таких ЭА-заместителей, как карбонильная группа (–С(О)–) и кетониминная группа (–С(NН)–). Ионизация гидроксигруппы и меркаптогруппы происходит в щелочной среде. У атомов кислорода и серы появляется еще одна неподеленная пара электронов, и они приобретают отрицательный заряд. Превращение нейтральной молекулы в анион значительно усиливает электронодонорность этих групп. Если эти группы входят в состав молекулы красителя, при ионизации происходит сдвиг λмакс в длинноволновую часть спектра и увеличение интенсивности поглощения. Ионизация аминогруппы происходит в кислой среде и заключается в присоединении протона к атому азота за счет его неподеленной пары электронов, при этом нейтральная молекула превращается в катион NH4+. В результате исчезновения неподеленной пары электронов аминогруппа перестает быть ЭД-заместителем; и молекула взаимодействует со светом так, как будто этого заместителя вообще нет. Ионизация ЭА-групп происходит в кислой среде и заключается в присоединении протона к атому кислорода или азота за счет имеющихся у них неподеленных пар электронов, при этом нейтральная молекула превращается в катион. Возникновение у ЭА-заместителей эффективного положительного заряда усиливает их электроноакцепторное действие, т. к. увеличивает способность групп –С(О)– и –С(NH)– притягивать электроны. В молекулах красителей эти свойства заместителей приводят к углублению цвета и усилению интенсивности окраски. В молекулах соединений, содержащих как ЭД-, так и ЭА-заместители, возможность ионизации увеличивается и соответственно возрастает зависимость окраски от рН среды. Большое влияние на поглощение света органическими соединениями оказывает пространственное расположение атомов в их молекулах. Если молекула расположена в одной плоскости (копланарна), то происходит перекрывание облаков π-электронов, облегчается их смещение по цепочке сопряженных двойных связей. Нарушение плоскостности молекулы затрудняет взаимодействие π-электронов. Для перевода такой молекулы в возбужденное состояние требуются дополнительные энергетические затраты для восстановления плоской структуры, вследствие чего возрастает уровень энергии молекулы в возбужденном состоянии, а следовательно, и энергия возбуждения, что сказывается на поглощении света: максимум поглощения сдвигается в сторону более коротких волн. На цвет влияет также присутствие в молекуле красителя иона металла. Он связывается с кислородом гидроксигруппы или азотом аминогруппы, замещая атом водорода, а с карбонильным кислородом или азотом в азогруппе, имеющим неподеленную пару электронов, образует координационную (донорно-акцепторную) связь. Поскольку атомы кислорода или азота, отдающие свою неподеленную пару электронов металлу, включены в цепочку сопряженных связей, то изменение их электронных оболочек приводит к изменению поглощения света. Таким образом, ответственной за цвет красителя является его хромофорная система. Ее основой в простых случаях является достаточно длинная цепь сопряженных двойных связей, а в более сложных — несколько (две и больше) изолированных, конкурирующих или перекрещивающихся цепей сопряженных двойных связей в составе единой молекулы. В хромофорную систему входят все присоединенные к сопряженным цепям ЭД- и ЭА-заместители и заместители, усиливающие или ослабляющие их электронодонорность и электроноакцепторность, а также комплексообразующие заместители и атомы металлов-комплексообразователей. Изложенные выше положения необходимо учитывать при выборе пищевых красителей, усиливающих или восстанавливающих (сохраняющих) цвет продукта. Часть используемых в пищевой промышленности красителей имеют природное происхождение. Они не обладают токсичностью, но для некоторых из них установлены допустимые суточные дозы (ДСД). Некоторые натуральные красители, обладая биологической активностью, одновременно являются вкусовыми и ароматическими веществами. К важнейшим натуральным красителям относятся кармин Е120 (красный), алканнин Е103 (красно-бордовый), куркумин Е100, рибофлавин Е101 (жёлтый), энокраситель Е163ii (красный или синий оттенки), сахарный колер Е150 (коричневый). В середине 19 века развитие химической промышленности привело к появлению большого количества синтетических красителей, которые по своим характеристикам (интенсивность окраски, свето-, кислото- и термостойкость) превосходили натуральные. В результате произошло снижение спроса на натуральные красители. В последние десятилетия наблюдается увеличение интереса к натуральным пищевым красителям. Это связано как с жесткой регламентацией использования синтетических красителей, так и со стремлением производителей придать пищевым продуктам статус натуральных. Лидирующее положение в объемах продаж занимают красные красители (около половины всего объема), затем идут желтые, оранжевые и зеленые. В большинстве случаев источником натуральных красителей является растительное сырье, в том числе отходы переработки овощей и фруктов. Одним из немногих исключений является кармин, выделяемый из тел самок насекомого кошениль. Качество натуральных красителей зависит от условий развития растений и животных (географического положения, климата, почв, питания), времени сбора растительного сырья, а также от технологии извлечения красящих веществ. Основным способом извлечения красящих веществ из природных объектов является экстракция растворителем, последующая очистка экстракта от сопутствующих соединений и стабилизация пигмента. В качестве растворителя-экстрагента используются этиловый спирт, вода, растительное масло и др. Основными веществами, обуславливающими окраску растительного сырья, являются каротиноиды (желтый, оранжевый и красный цвета), антоцианы (красный, фиолетовый и синий цвета) и хлорофиллы (зеленый цвет). Каротиноиды (a-, b-каротины, ликопин, капсорубин, лютеин и др.) выделяют из моркови, плодов шиповника, перца. Каротиноиды являются тетратерпенами и тетратерпеноидами (см. раздел Аромат) и включают две основные группы – каротины (см. лабораторную работу №1) и ксантофиллы (гидроксилированные каротиноиды). Их используют для окраски пищевых продуктов в желтый и желто-оранжевый цвета. К несомненным достоинствам натуральных красителей этой группы относится то обстоятельство, что некоторые из них проявляют А-витаминную активность (b-каротин, экстракт паприки, ликопин). Наиболее известным красителем из этой группы является b-каротин, широко применяемый в масложировой, молочной, макаронной и других отраслях промышленности. В последнее время наблюдается увеличение интереса к экстракту аннато, который характеризуется более высокой по сравнению с b-каротином светостойкостью. Антоцианы являются гликозидами, содержащими в качестве агликона-антоцианидина гидрокси- и метоксизамещённые соли флавилия (2-фенилхроменилия), у некоторых антоцианов гидроксильные группы ацетилированы (табл.9). В качестве углеводного остатка могут выступать моносахариды глюкоза, рамноза, галактоза, а также ди- и трисахариды. В настоящее время известно 20 антоцианидинов. Число же их возможных гликозидов и ацилированных гликозидов чрезвычайно велико. Антоцианы являются антиоксидантами и проявляют P-витаминную активность. Они содержатся в ягодах (чернике, малине, черной смородине и др.), овощах (баклажанах, свекле), красном вине.
Таблица 9.-Агликоны наиболее распространенных природных антоцианов
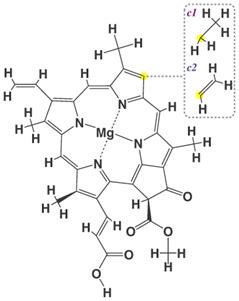
Натуральный магний-содержащий пигмент хлорофилл присутствует в листьях многих растений и обусловливает их зеленую окраску. Однако из-за низкой термостабильности природного хлорофилла применение в качестве натурального красителя нашли его медные комплексы.
Хлорофиллы с1 и с2
Действующими в России “Санитарными правилами по применению пищевых добавок № 1923-03” для большинства натуральных пищевых красителей не установлено максимально допустимых уровней содержания в пищевых продуктах. Исключение составляют лишь b-каротин (до 6 мг/ кг продукта в пересчете на каротин) и экстракт аннато (до 1600 мг/ кг продукта). В ряду натуральных красителей отдельного упоминания заслуживают меланоидины. Под меланоидинообразованием понимают взаимодействие восстанавливающих сахаров (моноз и восстанавливающих дисахаридов, как содержащихся в продукте, так и образующихся при гидролизе более сложных углеводов) с аминокислотами, пептидами и белками, приводящее к образованию темно-окрашенных продуктов — меланоидннов (от греч. меланос — темный). Этот процесс получил название реакции Майяра, по фамилии ученого, который в 1912 г. впервые его описал. Характерные ее признаки — потемнение продукта в результате образования трудно- или нерастворимых в воде темно-окрашенных соединений, снижение содержания редуцирующих сахаров и азота аминных групп, появление ароматообразующих веществ. Меланоидинообразование представляет собой окислительно-восстановительный процесс, состоящий из ряда последовательных и параллельных реакций. Механизм его сложен, реакция сопровождается образованием большого числа промежуточных продуктов, которые на следующих этапах взаимодействуют между собой и с исходными веществами. Скорость и глубина меланоидинообразования зависит от состава взаимодействующих продуктов, соотношения отдельных компонентов, рН среды, температуры, влажности. Активность аминокислот и сахаров в реакции Майяра снижается в следующих рядах: аминокислоты: лизин > глицин > метионин > аланин > валин > > глутамин > фенилаланин > цистин > тирозин; сахара: ксилоза > арабиноза > глюкоза > лактоза > мальтоза > фруктоза.
Наиболее интенсивно меланоидинообразование протекает в нейтральной и щелочной средах, легче проходит в концентрированных растворах, тормозится NaHSO3, H2SO4, Н2О2 и некоторыми другими соединениями. Образующиеся в ходе меланоидинообразования из аминокислот и сахаров карбонилсодержащие соединения (фурфурол, оксиметилфурфурол, ацетальдегид, изовалериановый альдегид, диацетил и др.) принимают участие в формировании аромата и вкуса готовых продуктов. К сожалению, использование натуральных красителей в пищевой промышленности сдерживается их низкой светостойкостью, невысокой устойчивостью к воздействию окислителей, недостаточной термостойкостью, а также невысокой красящей способностью. В связи с этим все большее распространение получают синтетические пищевые красители. Синтетические красители обладают значительными технологическими преимуществами по сравнению с натуральными. Они менее чувствительны к условиям технологической обработки и хранения и дают яркие, легко воспроизводимые цвета. Их себестоимость гораздо ниже себестоимости натуральных красителей, а производство не зависит от сезонности. Они обладают высокой красящей способностью, высокой устойчивостью к свету, окислителям и восстановителям, изменениям рН. Синтетические красители термостабильны, поэтому окрашенный продукт можно подвергать всем необходимым технологическим операциям, в т.ч. пастеризации, стерилизации, охлаждению и замораживанию. Без синтетических красителей современное многообразие и объемы выработки продуктов питания были бы существенно ограничены. С химической точки зрения они подразделяются на 5 классов: азокрасители (тартразин Е102, желтый 'солнечный закат' Е110, кармуазин Е122, пунцовый 4R Е124, черный блестящий Е151), триарилметановые (синий патентованный V Е131, синий блестящий Е133, зеленый S Е142, коричневый FK Е154, коричневый НТ Е155), ксантановые (эритрозин Е127), хинолиновые (хинолиновый желтый Е104) и индигоидные (индигокармин Е132). Препараты синтетических красителей содержат, как правило, 80-85% основного красителя, но могут также изготавливаться с наполнителем (солью или сахаром). Иногда в продаже встречаются водные растворы красителей. Такие 'разбавленные' красители применяются в более высокой дозировке, чем порошковые. Кроме того, они имеют ограниченный срок годности. Часть пищевых красителей растворяется в жирах или в спирте, многие образуют нерастворимые комплексы (лаки) с ионами металлов и в такой форме в качестве пигментов используются для окрашивания порошкообразных продуктов, драже, таблеток, жевательной резинки. Свойства красителей определяют возможность их применения в тех или иных пищевых продуктах. Синтетические пищевые красители применяются как индивидуально, так и в смесях друг с другом. Смеси красителей используются для получения цветов и оттенков, которых не удается добиться при применении индивидуальных красителей. Обычная рекомендуемая доза синтетических красителей – 10-50 г на тонну готового пищевого красителя. Максимально разрешённая дозировка синтетических красителей - 100 г/т. Красители используются в кондитерской промышленности, при изготовлении напитков, маргарина, некоторых видов консервов. Существует перечень продуктов, в которых не допускается использование пищевых красителей.
Лабораторные работы 1. Получение кристаллического β-каротина из моркови
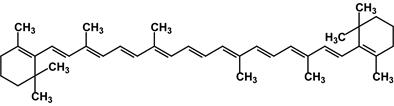
β-Каротин (Е-160a) представляет собой жёлто-оранжевый краситель; он является источником витамина А, антиоксидантом, антиканцерогеном.
b-каротин β-Каротин содержится в растительных пищевых продуктах: моркови, шпинате, салате, петрушке, зелёном луке, щавеле, красном перце, чёрной смородине, чернике, крыжовнике, персиках, абрикосах. β-Каротин получают синтетическим (в том числе микробиологическим) путем или выделяют из природных источников, например из криля, в смеси с другими каротиноидами в виде водо- или жирорастворимых форм. Он применяется для окрашивания и витаминизации маргаринов, майонезов, кондитерских и хлебобулочных изделий, безалкогольных напитков, для подкрашивания и витаминизации молочных консервов, сгущенного молока, сливок, сыра, творога, жидких и пастообразных молочных продуктов. Оборудование: Мешалка Стакан на 200 мл Водяная баня Центрифуга Воронка Бюхнера Одногорлая колба Фильтры Ход работы: Получение коагулята. Хорошо вымытую свежую морковь пропускают через мясорубку. Мезгу частями переносят на кусок хлопчатобумажной ткани и из каждой порции отжимают сок руками. Отжатую мезгу помещают в стакан, добавляют туда 100 мл воды и 40 минут интенсивно перемешивают механической мешалкой. После этого процеживают раствор и снова отжимают сок. Объединив обе порции сока и раствор, проводят термическую коагуляцию белковых веществ (см. примечание 1). Для этого колбу с соком нагревают на водяной бане, слабо помешивая, и выдерживают некоторое время при 60—70 °С (см. примечание 2). Нагретому соку дают медленно отстояться. По достижении комнатной температуры сок быстро расслаивается: основная масса белка оседает на дно колбы, меньшая часть сосредотачивается на поверхности жидкости. Прозрачный раствор между этими слоями осторожно сифонируют с помощью изогнутой стеклянной трубочки, регулируя ее уровень так, чтобы можно было слить максимальное количество жидкости. Остальное центрифугируют (см. примечание 3). Выход влажного коагулята 30-40 г. Получение кристаллического каротина.Свежеосажденный коагулят переносят в делительную воронку, хорошо взбалтывают с 1 г порошкообразного едкого натра и 10 мл четыреххлористого углерода (см. примечание 4). Экстрагирование проводят 4 раза. От примесей белков и щелочи объединенный раствор очищают промывкой небольшим количеством дистиллированной воды и отгоняют под вакуумом в токе инертного газа при 30 – 40 °С. В колбу с теплым маслообразным остатком прибавляют небольшими порциями 3 г отмученного и хорошо просушенного при 100 °С кизельгура. Колбу встряхивают около часа, пока масса не перестанет прилипать к стенкам. Чтобы удалить из адсорбата неомыляемые жироподобные примеси, его переносят в воронку на стеклянный фильтр и перемешивают с порцией петролейного эфира (т. кип. 50—60 °С). Фильтрование проводят не под вакуумом, а под небольшим давлением инертного газа. Для этого воронку закрывают пробкой, в которую вставлена трубка. Трубку соединяют с камерой футбольного мяча или кислородной подушкой, заполненной углекислым газом или азотом. Адсорбат промывают дважды. Осадок переносят в колбу с пришлифованным обратным холодильником, заливают 15 мл этилового спирта и кипятят 2—3 минуты. Горячую смесь фильтруют через тот же фильтр, но уже применяя отсасывание. Такой промывкой, повторяющейся дважды, удаляют стерины. Снимают каротин с адсорбента несколькими порциями хлороформа прямо на воронке. Для этого перед всасыванием очередной части раствора осадок тщательно перемешивают с каждым новым количеством растворителя. Хлороформ отгоняют под вакуумом при 30 °С досуха. Остаток снова растворяют, нагревая в 1 мл хлороформа. К раствору быстро добавляют 20 мл кипящего этилового спирта. Выпавшие кристаллы каротина тут же отфильтровывают на стеклянном фильтре № 2, промывают горячим метиловым спиртом, вновь растворяют в 1 мл хлороформа и осаждают 20 мл кипящего этилового спирта. Полученный кристаллический каротин высушивают в вакуум-эксикаторе. Выход 0,03 - 0,04 г (см. примечание 5). Чистый β-каротин — кристаллы темно-красного цвета с фиолетовым оттенком и металлическим блеском; т. пл. 176-177 °С. Для идентификации α-, β- и γ-каротинов целесообразно провести качественное хроматографирование. Немного каротина (0,01 г) растворяют в минимальном количестве петролейного эфира и хроматографируют на небольшой колонке. Колонку заполняют окисью магния, активированной нагреванием при 100°С. После промывания тем же растворителем на колонке выявляются три характерные зоны. Отсутствие других окрашенных зон свидетельствует о достаточной чистоте полученного препарата.
Примечания. 1. Во время коагуляции практически весь каротин, имеющийся в соке, адсорбируется на выпавших в осадок веществах и может быть таким путем сконцентрирован. 2. Нагревание выше 70 °С вызывает побурение белковой массы и сопровождается значительной потерей каротина. 3. Применение фильтров разных систем в данном случае себя не оправдывает, так как вязкая белковая масса забивает все поры фильтра. 4. Липоидная фракция белкового коагулята в последующей экстракции загрязняет каротин и затрудняет его кристаллизацию, поэтому проводится щелочное омыление. Если совместить омыление с экстракцией, соли жирных кислот прочно адсорбируются белком и не образуют стойкой эмульсии. 5. Чтобы выход был больше, все операции по очистке каротина необходимо вести в условиях, по возможности исключающих влияние света и воздуха. Контрольные вопросы: 1. Какие натуральные красители оранжевого цвета вы знаете? 2. Чем обусловлена относительно низкая устойчивость каротинов? 3. В состав каких пищевых продуктов входит β-каротин?

Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2024 stydopedia.ru Все материалы защищены законодательством РФ.
|