
|
|
ГОМОФУНКЦИОНАЛЬНЫЕ УГЛЕВОДОРОДЫУглеводород образует функциональные производные, если один или более атомов водорода в нем замещается на другой атом или группу атомов, называемых функциональной группой. ГАЛОГЕНОПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ К галогенопроизводным углеводородов относят продукты замещения одного или нескольких атомов водорода в молекуле на галоген. По степени замещения различают: · моногалогенопроизводные; · дигалогенопроизводные: геминальные (hemi – близнецы) – оба атома галогена находятся при одном углеродном атоме; вицинальные – атомы галогенов находятся у соседних углеродных атомов; · полигалогенопроизводные. По строению углеводородного радикала: · алкил галогениды; · алкенилгалогениды; · аллилгалогениды; · арилгалогениды; · бензилгалогениды. Из галогенопроизводных непредельного ряда теоретический и практический интерес представляют соединения двух типов: · винилхлорида – атом галогена находится рядом с кратной связью; · аллилхлорида – атом галогена отделен от кратной связи одним метиленовым звеном. По типу атома углерода, соединенного с галогеном: · галогенпроизводные со связью С(sp3)–Hal · галогенпроизводные со связью С(sp2)–Hal · галогенпроизводные со связью С(sp)–Hal Соединения, в которых двойная связь и атом галогена далеко отстоят друг от друга, химически не отличаются от обычных алкенов и галогенопроизводных насыщенных углеводородов. В зависимости от того, с каким атомом углерода связан галоген выделяют: · первичные; · вторичные; · третичные. В зависимости от природы галогена: · фтористые; · хлористые; · бромистые; · йодистые галогенопроизводные. ГАЛОГЕНПРОИЗВОДНЫЕ АЛИФАТИЧЕСКОГО РЯДА Номенклатура. Для названия галогенпроизводных алифатического ряда используют следующие номенклатуры: · тривиальную; · радикально-функциональную; · систематическую. Тривиальная. Некоторые галогенпроизводные алифатического ряда сохранили тривиальные названия: хлороформ (CHCl3), йодоформ (CHI3). Радикально-функциональная используется для соединений с простым углеводородным скелетом и соответственно легко называемым радикалом (часть 1, табл. 8) с указанием типа атома углерода. Систематическая (заместительная, международная, ИЮПАК). Название строится по общим правилам, т. е. с учетом старшинства функциональной группы над алкильными радикалами. Меньший локант получает заместитель, который в названии пишется первым, а сами заместители перечисляются в алфавитном порядке. Изомерия. Для галогенпроизводных алифатического ряда характерны следующие виды изомерии: Структурная: · скелетная; · изомерия положения. Пространственная: · конформационная; · оптическая. Способы получения. Природные источники галогенпроизводных алифатического ряда неизвестны, поэтому все эти соединения получают синтетически. Способы получения могут быть классифицированы по нескольким признакам: · по природе исходного субстрата; · по природе галогенирующего агента; · по реакции, используемой для синтеза. 1. Реакции замещения. 1.1 Прямое галогенирование алканов (часть 1, глава 8.1). 1.2 Реакции замещенияатома кислорода в карбонильных соединениях, карбоновых кислотах и спиртах и т. д. 1.2.1 Получение из спиртов. Проба Лукаса:
1.2.2 Взаимодействие с галогенангидридами миниральных кислот. К эффективным реагентам относятся галогениды фосфора и серы PCl3, POCl3, PCl5, PBr3, PBr5, PI3, SOCl2, SF4.
1.2.3 Получение из карбонильных соединений:
Получение из карбоновых кислот:
2. Взаимное замещение атомов галогенов (Финкельштейн, 1910 г.):
3. Галогенметилирование аренов:
4. Реакция декарбоксилирования карбоновых кислот(реакция Хунсдикера): 4.1
4.2
5. Реакции присоединения к непредельным углеводородам (часть 1, главы 8.2–8.4) Основные физические параметры связи С–Наl.У галогенпроизводных углеводородов появляется новый тип связи С–Наl, свойства которой определяют реакционную способность этого класса соединений. Галоген образует с атомом углерода простую σ-связь, которая в отличии от неполярных или слабополярных С–С и С–Н связей, как правило, заметно поляризована, и электронная плотность смещена к более электроотрицательному атому галогена:
Очевидно, что большинство реакций галогенопроизводных связано с разрывом связи С–Наl. Основные характеристики связей С–Наl в сравнении со связью C(sp3)–H приведены в табл.1.
Таблица 1 – Физические характеристики связи С–Hal
Данные таблицы 1 свидетельствуют, что связи С–Наl имеют значительные дипольные моменты, т. е. они полярны. Вследствие большей электроотрицательности атома галогена по сравнению с атомом углерода ковалентная связь С–Hal поляризована. Дипольный момент связей мало изменяется при переходе от одного галогена к другому, так как длина связи С–На1 растет, а электроотрицательность галогена, определяющая величину заряда, падает. Поляризуемость связи С–Hal существенно зависит от природы галогена и закономерно увеличивается в ряду от C–F к C–I. Значительную поляризуемость связи C–I можно объяснить тем, что в атомах с большими атомными радиусами внешние электроны слабее связаны с ядром и поэтому более чувствительны к влиянию внешнего электрического поля. На полярность связи влияют: · природа атома галогена; · величина и строение углеводородного радикала. Большинство реакций галогеналканов, широко используемых в органическом синтезе, связано с нуклеофильной атакой электрондефицитного атома углерода и замещением галогена на остаток нуклеофила. В подобных реакциях наименее реакционноспособными оказываются фторпроизводные. Атом фтора компактен благодаря маленькому ковалентному радиусу, при этом связь С–F наиболее полярная, самая короткая и прочная, поэтому у фторпроизводных легче разорвать связь С–С, нежели С–F. Наибольшую активность проявляют йодзамещенные, что объясняется наименьшей среди галогенов электроотрицательностью атома йода; его оболочка, будучи достаточно большой, легко деформируется под внешним воздействием, т. е. связь С–I легко поляризуется. Именно фактор поляризуемости оказывается решающим для химического поведения галогеналканов во многих реакциях. Химические свойства. Для галогеналканов характерны реакции нуклеофильного замещения, отщепления галогеноводородов (элиминирование) с образованием алкенов, взаимодействие с металлами (получение металлорганических соединений), восстановление (образование алканов). В органическом синтезе особенно широко распространены реакции замещения, позволяющие получать на основе галогеналканов самые разнообразные органические соединения. 1. Реакции нуклеофильного замещения.Галогеналканы способны взаимодействовать с самыми разнообразными нуклеофильными реагентами с замещением галогена на остаток нуклеофила:
где Nu– = OH–, NO2–, CN–, HOH, NR3, ROH, HS–, SCN–, N3, NH3, PR3–, SR2.
Протекание реакции зависит от строения исходного галогеналкана. Наиболее реакционноспособными являются третичные соединения, затем следуют вторичные, наименьшую активность проявляют первичные галоген-алканы. Основной причиной подобной закономерности является различие в механизмах реакций. Изучение скорости реакции гидролиза показало: · у первичных галогеналканов она зависит от концентрации обоих реагентов; · у третичных алкилгалогенидов скорость пропорциональна концентрации только галогеналкана. Это означает, что на лимитирующей стадии реакции у первичных галогеналканов участвуют две частицы (бимолекулярный механизмSN2), у третичных – участвует только одна молекула галогеналкана (мономолекулярный механизмSN1). Механизм реакции мономолекулярного нуклеофильного замещения (SN1), асинхронный механизм.Карбокатионный механизм характерен для третичных галогеналканов и является двухстадийным. На первой (лимитирующей) стадии полярная связь С–Hal, разрыхляясь под действием диполей растворителя, подвергается гетеролитическому распаду с образованием третичного карбокатиона:
На следующей стадии образовавшийся карбокатион, обладающий высокой реакционной способностью, вследствие своей выраженной электрофильности (шесть электронов у центрального атома углерода), быстро стабилизируется, взаимодействуя с окружающими его нуклеофильными частицами с образованием молекулы спирта:
В карбокатионе атом углерода находится в sp2 гибридном состоянии. Связи, образованные этим атомом, находятся в одной плоскости. Атака нуклеофила равновероятна с обеих сторон плоскости, то есть происходит рацемизация (уменьшение оптической активности). Соотношение R- и S-изомеров часто неэквивалентно и во многом зависит от типа растворителя. Иногда наблюдается преобладание продукта с наиболее обращенной конфигурацией, т. к. карбокатион и Hal – образуют ионную пару, и анион мешает атаке нуклеофила с фронта, т. е. со стороны, где он расположен. В водной среде реакция замещения обратима. В обратной реакции образовавшийся спирт протонируется, взаимодействуя с присутствующим в среде протоном и, отщепляя воду, превращается в карбокатион, который далее может соединяться с Hal –. Строение алкилгалогенидов существенно влияет на поведение их в SN1 реакциях, т. к. лимитирующей стадией реакции является образование карбокатиона, следовательно, чем устойчивее карбокатион, тем легче протекает реакция SN1:
третичный > вторичный > первичный
т. е. стабилизирующее влияние алкильных радикалов, связанных с заряженным атомом углерода, падает, поэтому скорость реакции соответствующих галогеналканов в SN1 реакциях уменьшается при переходе от третичных к первичным. Механизм реакции бимолекулярного нуклеофильного замещения (SN2).На промежуточной стадии возникает так называемое переходное состояние, или активированный комплекс, в котором новая связь еще не образовалась, а старая еще не разрушилась:
Связь С–Нal рвется в момент завершения образования новой связи С–О (синхронная реакция). При этом часть энергии, необходимой для разрыва связи С–Hal, возмещается за счет энергии образования новой связи С–О. Расчет показывает, что наименьшая затрата энергии требуется в случае, когда приближение атакующего нуклеофила (OH–) происходит не со стороны атома галогена (мешает дробный одноименный заряд), а с тыла, и атака осуществляется по линии, соединяющей центры атомов углерода и галогена. Атакуемый атом углерода в переходном состоянии меняет тип гибридизации, приобретая почти sp2 гибридизацию. Поэтому связанные с ним атомы расположены планарно в плоскости, перпендикулярной линии атаки. В переходном состоянии р-орбиталь атома углерода перекрывается одновременно с орбиталями атакующего гидроксила и уходящего галогена. Однако это не настоящие σ-связи, новая связь С–О еще не образовалась, а старая С–Hal разрыхлена. Скорость SN2 реакций связана со строением радикала и уменьшается с увеличением разветвленности при атакуемом атоме углерода или у соседнего с ним атома. Увеличение числа заместителей вокруг реакционного центра приводит к возникновению пространственных препятствий во взаимодействии с нуклеофилом, затрудняя его подход к атакуемому атому и образование частичной связи с ним, поэтому скорость SN2 процесса резко падает. В третичных производных экранирование алкильными радикалами реакционного центра настолько велико, что они практически не вступают в реакции SN2. Факторы, влияющие на ход нуклеофильного замещения Механизм Sn реакций и их скорость зависят от следующих факторов: · строения субстрата; · природы уходящей группы; · природы растворителя; · нуклеофильности реагента. А. Влияние строения субстрата.Строение субстрата оказывает существенное влияние на реакционную способность соединений в реакциях нуклеофильного замещения. С точки зрения реакционной способности, связанной с подвижностью галогена в Sn реакциях, все галогенопроизводные углеводородов можно разделить на три типа: · с нормальной реакционной способностью; · с пониженной реакционной способностью; · с повышеннойреакционной способностью. К первой группе относятся галогенопроизводные алканов, циклоалканов, а также непредельных и ароматических углеводородов, в которых галоген находится в β- и далее положении по отношению к π-электронам:
Ко второй группе относятся галогенопроизводные алкенов и ароматических углеводородов, в которых атом галогена непосредственно связан с одним из атомов углерода двойной связи, т. е. в которых имеется связь С(sр2)–На1:
Галогенопроизводные этого типа очень инертны в Sn реакциях, атом галогена замещается с трудом в жестких условиях. Причина пониженной реакционной способности соединений такого типа в Sn реакциях заключается во взаимодействии НЭП атома галогена с π-электронами двойной связи:
К галогенпроизводным с повышенной подвижностью галогена в Sn реакциях относятся все соединения, содержащие галоген в α-положении к кратным связям:
Соединения этой группы очень активны в реакциях Sn , они протекают быстрее и легче, чем у галогенопроизводных с нормальной реакционной способностью. Причина подобного различия обусловлена пониженной энергией диссоциации связи С–Hal у галогенидов такого типа по сравнению с остальными двумя группами галогенпроизводных. Снижение энергии диссоциации связи обусловлено повышением стабильности карбкатионов, образующихся при разрыве связи С–Hal:
Карбкатионы аллильного и бензильного типа очень устойчивы вследствие значительной делокализации положительного заряда с участием π-электронов кратной связи или бензольного кольца. Природа углеводородного радикала, связанного с уходящей группой, существенно влияет не только на активность субстрата в нуклеофильных реакциях, но и на предпочтительность реализации замещения по тому или иному механизму. Чтобы оценить склонность субстрата к взаимодействию по механизму Sn2 или Sn1 необходимо учитывать: · пространственное строение заместителя; · энергию диссоциации связи С-уходящая группа. Доминирующим фактором в случае реакции Sn2 является стерический фактор: большие по объему заместители при С-атоме, связанным с уходящей группой, уменьшают реакционную способность субстрата: · затруднена атака с тыла, так как объемные заместители заслоняют атакуемый атом углерода; · возникают большие стерические препятствия в переходном состоянии: атакуемый С-атом окружен пятью заместителями, и если три из них занимают большой объем, то образование переходного состояния затруднено. В соответствии с этим тенденция к Sn2 реакциям и их скорость увеличивается в ряду от третичных к вторичным и первичным субстратам. Sn1 реакции могут протекать лишь у тех субстратов, у которых связь С–Hal имеет низкую энергию диссоциации, так как в этом случае на лимитирующей стадии происходит предварительная диссоциация этой связи. Энергия диссоциации связи тем ниже, чем стабильнее образующийся карбокатион, устойчивость которого зависит от степени делокализации положительного заряда:
С+Н2СН3 < С+Н(СН3)2 < С+(СН3)3
Так же как и соответствующие свободные радикалы, карбокатионы аллильного или бензильного типа очень устойчивы вследствие значительной делокализации положительного заряда с участием π-электронов кратной связи или бензольного кольца. Таким образом, третичные субстраты обычно вступают в реакции SN1, а для первичных предпочтителен SN2 механизм. Субстраты вторичные, а также бензильного и аллильного типа могут реагировать по SN2 и SN1 в зависимости от природы растворителя. Б. Влияние природы уходящей группы (нуклеофила). Гетеролитический разрыв связи С–Hal протекает тем легче, чем беднее энергией уходящая группа, т. е. является устойчивым ионом или нейтральной молекулой. Плохие уходящие группы богаты энергией; в них невелика или отсутствует степень делокализации отрицательного заряда. В ряду алкилгалогенидов реакционная способность в реакциях нуклеофильного замещения изменяется следующим образом:
R–F < R–Cl < R–Br < R–I
Самыми активными при прочих равных условиях являются алкилйодиды, так как I самая выгодная уходящая группа. Анион йода имеет большой объем, отрицательный заряд распределяется на большем пространстве, что уменьшает энергию аниона, т. е. повышает его стабильность. Устойчивость уходящей группы обуславливает, в свою очередь, низкую энергию диссоциации связи С–На1, т. е. в ряду C–F, C–C1, С–Вr, C–I энергия диссоциации связи уменьшается. В. Влияние природы растворителя.Реакции нуклеофильного замещения, будучи ионными, протекают только в растворах и в значительной мере зависят от характера растворителя. Полярный растворитель принимает участие: · в ионизации молекул исходных веществ; · в стабилизации образующихся ионов за счет их сольватации; · в стабилизации переходного состояния. По сольватирующей способности растворители подразделяют на: · протонные; · апротонные. К протоннымотносятся легко ионизируемые растворители, содержащие подвижные атомы водорода, такие как вода, аммиак, спирты, муравьиная и уксусная кислоты, этиленгликоль. Эти растворители за счет НЭП гетероатомов хорошо сольватируют катионы, а благодаря способности образовывать водородные связи также сольватируют и анионы. Поэтому они способствуют протеканию реакций по механизму Sn1, где образуются и те и другие ионные частицы. К апротонным полярнымрастворителям относятся: ацетон, диоксан, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), диэтиловый эфир. Апротонные полярные растворители за счет НЭП гетероатомов сольватируют катионы и нуклеофил оказывается «обнаженным», сохраняя свою активность по отношению к субстрату. Однако поскольку апротонные полярные растворители не способны к образованию водородных связей, то оставляют анионы несольватированными, в результате облегчается протекание реакций, где нуклеофил принимает участие в лимитирующей стадии процесса, т. е. Sn2. Г. Влияние нуклеофильности реагента. Нуклеофильность частицы является мерой того, насколько быстро она может реагировать в реакциях нуклеофильного замещения с положительно заряженным атомом углерода. Нуклеофильность зависит от следующих факторов: · основности реагента; · поляризуемости реагента; · природы растворителя. Нуклеофильность пропорциональна основности и поляризуемости, т. е. чем больше размер атома, тем выше его нуклеофильность. Это обусловлено тем, что при увеличении размера атома уменьшается притяжение между периферическими электронами и ядром, в результате чего электроны становятся более подвижными и поэтому легче образуют связь с положительно заряженным реакционным центром субстрата. Такие легкополяризуемые реагенты, обладают высокой нуклеофильностью в растворителях различного типа. Существенное влияние на нуклеофильность слабополяризуемых ионов оказывает природа растворителя. В полярных растворителях ионы находятся в сольватированном состоянии, и чем сильнее сольватирован анион, тем меньше его нуклеофильность. Анион-нуклеофил может атаковать атом углерода субстрата только после удаления сольватной оболочки, т. е. когда он «обнаженный» (несольватированный). В больших анионах отрицательный заряд делокализован (распределен) в сравнительно большом объеме и степень сольватации, прочность водородных связей меньше. Поэтому больший анион освобождается от растворителя с меньшей затратой энергии. В протонных растворителях большие по размеру анионы являются более сильными нуклеофилами. Сильное взаимодействие между растворителем и анионом малого размера подавляет его нуклеофильность. Реагенты по возрастанию их нуклеофильности в SN2 реакциях в протонных растворителях можно расположить таким образом: F– < Н2О < С1– < С6Н5О– < Вr– < ОН– < OR– < NH3 < CN– < I– нуклеофильность возрастает с увеличением поляризуемости. Апротонный растворитель не способен сольватировать анионы за счет образования водородных связей с ними, поэтому анионы в этом случае несольватированные. Следовательно, анион малого размера, имеющий большой «–» заряд, является более сильным нуклеофилом.
Таблица 2 – Сравнение SN1 и SN2 реакций
Особенности протекания реакций нуклеофильного замещения с амбидентными нуклеофилами. Правило Корнблюма.Нуклеофил, содержащий два реакционных центра, называется амбидентным.Типичным амбидентным ионом является нитрит-ион, который имеет два нуклеофильных центра – «N» и «О»:
Реакция нуклеофильного замещения с участием амбидентного нуклеофила цианид-иона будет давать нитрилы (цианиды) или изонитрилы (изоцианиды) в зависимости от того, какой атом является атакующим – углерод или азот:
Направление реакции с участием амбидентных нуклеофилов зависит от ее механизма (правило Корнблюма): · в SN2 реакциях атакующим атомом амбидентного нуклеофила будет тот, который более нуклеофилен, т. е. имеет большую поляризуемость; · в SN1 реакциях с карбокатионом взаимодействует реакционный центр амбидентного иона с более высокой электронной плотностью, т. е. атом с наибольшей электроотрицательностью. 2. Аллильная перегруппировка. В случае реакции SN в аллильных соединениях обычно наблюдается аллильная перегруппировка:
Если превращение осуществляется по SN1, наиболее характерному для таких систем, то в результате реакции образуется продукт замещения и продукт аллильной перегруппировки практически в равных количествах. На лимитирующей стадии реакции образуется аллильный карбокатион, в котором делокализация «+» заряда. Атака нуклеофила на второй стадии практически равновероятна по 1 и 3 углеродному атому аллильной системы:
В случае реализации замещения по SN2 образуется только продукт аллильной перегруппировки, т. к. атака нуклеофила осуществляется по 3 углеродному атому аллильной системы:
3. Реакции отщепления (элиминирования). Каждый нуклеофильный реагент является основанием, способным отщеплять протон от субстратов с подвижными атомами водорода, поэтому параллельно с реакцией SN протекает реакция ЕN:
Этот тип превращения относится к реакциям 1,2- или β-элиминирования. Атом углерода, от которого отщепляется галоген, называют α-углеродным атомом, а соседний атом углерода, теряющий протон β-углеродным атомом. Реакции α-элиминирования: · атомы галогена и водорода отщепляются от одного и того же углеродного атома, что приводит к образованию карбена. В качестве примера можно привести гидролиз хлороформа под действием сильного основания:
· при отсутствии β-водородных атомов и действии сильных нуклеофилов:
Механизм ЕN2. В молекуле алкилгалогенида влияние отрицательного индуктивного эффекта атома галогена передается по σ-связям к β-водородным атомам, вследствие чего они приобретают большую, чем в незамещенных алканах, способность отщепляться в виде протона. Нуклеофил принимает участие в образовании переходного состояния, атакуя атом водорода у β-углеродного атома и отрывает его в виде протона. Одновременно с этим процессом происходит образование двойной связи и вытеснение уходящей группы в виде аниона:
Механизм ЕN1. Реакция протекает как двухстадийный процесс:
На первой стадии медленно образуется карбокатион (лимитирующая стадия), а затем нуклеофил отрывает протон у β-углеродного атома:
Направление и стереохимия реакций элиминирования.В реакциях отщепления галогеноводородов с участием несимметричных вторичных и третичных алкилгалогенидов возможно образование алкенов различного строения:
Для предсказания результата реакции элиминирования галогеноводорода существует эмпирическое правило Зайцева,которое гласит:водород отщепляется преимущественно от соседнего наименее гидрогенизированного β-углеродного атома.Это же правило может быть сформулировано иначе: дегидрогалогенирование алкилгалогенидов протекает с образованием алкенов, содержащих наибольшее число алкильных групп у двойной связи. В приведенной выше реакции, согласно правилу Зайцева, предпочтительно направление 2, т. е. образование бутена-2. Правило Зайцева легко интерпретируется, учитывая относительную стабильность образующихся алкенов: алкен с большим числом алкильных групп у двойной связи наиболее термодинамически стабильный продукт реакции. Следует отметить, что не все реакции подчиняются правилу Зайцева. В тех случаях, когда при проведении реакции возникают какие-либо пространственные затруднения, это правило может не соблюдаться. Например, при отщеплении НВr от 2-бром-2,4,4-триметилпентана преимущественно образуется менее замещенный алкен (1), так как в более замещенном алкене (2) слишком велики пространственные затруднения, обусловленные взаимным отталкиванием трет-бутильной группы и находящейся в цис-положении по отношению к ней метильной группы:
Несоблюдение правила Зайцева имеет место и в тех случаях, когда возникают пространственные затруднения, связанные с размером основания – реагента. Так, при дегидробромировании 2-метил-2-бромбутана под действием алкоголятов различных спиртов возможно образование двух алкенов:
Факторы, влияющие на конкуренцию реакций SN и ЕN.Реакции нуклеофильного замещения галогена и реакции элиминирования протекают под действием одного и того же нуклеофильного реагента: его атака поуглеродному атому приводит к замещению, а атака по β-водородному атому – к элиминированию:
Следовательно, реакции SN и ЕN всегда конкурируют. Соотношение между ними зависит, главным образом, от следующих факторов: · основности реагента; · строения субстрата; · температуры реакции. А. Основность реагента, т. е. сродство к протону: чем выше основность нуклеофила, тем больший вклад реакции элиминирования. Некоторые наиболее распространенные реагенты по увеличению основности можно расположить в следующий ряд: –NH2 > –OC2H5 > –OH > –OC6H5 > Cl– > Br– > I– Если в реакции применять нуклеофил с достаточно высокой основностью, то можно добиться преимущественного протекания реакции элиминирования:
Б. Строение субстрата.На скорость дегидрогалогенирования влияет как природа галогена, так и строение углеводородного остатка алкилгалогенида. В зависимости от природы галогена при одинаковом R скорость отщепления HHal изменяется в следующем порядке, соответствующем прочности связи С–На1:
R–I > R–Br > R–Cl > R–F
Элиминирование происходит в большей степени при переходе от первичных к третичным алкилгалогенидам, у которых наиболее низкая энергия диссоциации связи С–На1. Кроме того, строение субстрата предопределяет и механизм реакции элиминирования. Как отмечалось выше, первичные субстраты реагируют по ЕN2 механизму, а для третичных алкилгалогенидов предпочтительнее ЕN1 реакция. В. Влияние температуры реакции.Повышение температуры всегда способствует большему вкладу реакции элиминирования, так как в системах с конкурирующими реакциями замещения и отщепления реакция элиминирования требует более высокой энергии активации. При элиминировании разрываются две связи в субстрате, а при замещении лишь одна. Более высокие энергетические требования удовлетворяются, когда реакция проводится при повышенных температурах. 3. Реакции галогенопроизводных с металлами. Алкил- и арилгалогениды реагируют с металлами с образованием металлоорганических соединений, в которых имеется связь углерод – металл. Из металлоорганических соединений металлов первой группы наиболее важными и хорошо изученными являются литий- и натрийорганические соединения. 3.1 Прямое металлирование галогеналканов в растворе диэтилового эфира или тетрагидрофурана приводит к образованию литийалкилов:
R–Hal + 2Li
Реакция проводится в атмосфере инертного газа, т. к. образующиеся продукты могут реагировать с кислородом, водой, диоксидом углерода и может происходить их самовоспламенение. 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2024 stydopedia.ru Все материалы защищены законодательством РФ.
|

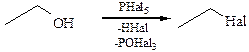



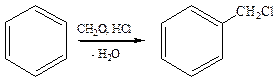

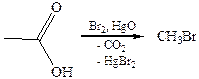






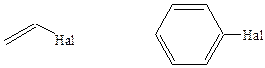


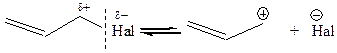






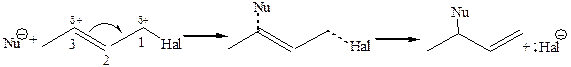










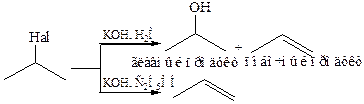
 R–Li + LiHal
R–Li + LiHal