
|
|
Б) Расчеты теплот сгоранияСтандартной теплотой сгорания называют теплоту выделяющегося при окислении одного моля вещества, взятого в стандартном состоянии при данной температуре, кислородом до конечных продуктов окисления. Для большинства органических соединений конечными продуктами окисления являются СО2, Н2О, SO2 ,N2. Из закона Гесса следует, что теплота химической реакции равна алгебраической сумме теплот сгорания реагирующих веществ с учетом их стехиометрических коэффициентов, причем теплоты сгорания продуктов реакции берут со знаком минус, а теплоты сгорания исходных веществ – со знаком плюс: DН реакции = SniDНсгор(исходн) - SniDНсгор.(прод.). (2.24) Стандартные теплоты образования используют обычно при вычислении теплот реакций с участием неорганических веществ, а стандартные теплоты сгорания – при расчете теплот реакций с участием органических веществ. в) Теплота нейтрализации Теплотой нейтрализации называют теплоту, необходимую для нейтрализации одного моль-эквивалента кислоты одним моль-эквивалентом основания. Теплота нейтрализации сильных кислот сильными основаниями не зависит от природы кислоты и основания, например: Na+ + OH- + H+ + Cl- = Na+ + Cl- + H2O; DН = - 57,30 кДж/моль; H+ + OH- = H2O; DН1 = -57,32 кДж/моль; К+ + OH- + H+ + Cl- = К+ + Cl- + H2O; DН2 = - 57,73 кДж/моль; H+ + OH- = H2O; DН2 = -57,73 кДж/моль. Тепловой эффект приведенных реакций практически одинаков и равен 57,52 кДж/моль. Такое постоянство легко объяснить тем, что сильные кислоты и основания в водных растворах практически полностью ионизированы и реакция нейтрализации сводится к взаимодействию ионов H+ и OH-: H+ + OH- = H2O; DН = -57,52 кДж/моль. В случае реакции нейтрализации слабых кислот и слабых оснований такого постоянства не наблюдается, так как часть тепла расходуется на ионизацию слабой кислоты или слабого основания. г) Теплота растворения В зависимости от природы растворенного вещества и природы растворителя процесс растворения сопровождается выделением или поглощением тепла. Мольной теплотой растворения называют количество тепла, выделяющееся и поглощающееся при растворении одного моля вещества в таком объеме растворителя, когда его дальнейшее прибавление не вызывает выделения или поглощения тепла. Обычно при определении теплоты растворения берется более чем 300‑кратный избыток растворителя. Теплота растворения твердых тел представляет собой теплоту разрушения кристаллической решетки DНр и теплоту сольватации DНс: DНраств. = DНр + DНс. Для разрушения кристаллической решетки необходимо затратить некоторое количество энергии, поэтому теплота этого процесса будет положительна. Сольватация всегда сопровождается выделением тепла и, следовательно, теплота этого процесса отрицательна. Отсюда следует, что теплота растворения может быть как положительной, так и отрицательной. Отрицательная теплота растворения указывает на то, что количество теплоты, выделяющееся при сольватации, больше, чем энергии, необходимой для разрушения кристаллической решетки. В случае положительной теплоты растворения количество энергии, необходимое для разрушения кристаллической решетки, больше энергии, которая выделяется при сольватации. В тех случаях, когда в качестве растворителя используют воду, теплота, которая выделяется при взаимодействии растворенного вещества с растворителем, называется теплотой гидратации. Теплоту гидратации, в соответствии с законом Гесса, рассчитывают по разности между теплотой растворения безводного соединения и теплотой растворения гидрата. Лекция 3. 3.1. Основной смысл и значение второго закона Все многообразие процессов, происходящих в окружающем нас мире – в природе, в производственных и других условиях, – можно разделить на три группы: 1) процессы, для совершения которых требуется затрата работы извне в количестве, прямо пропорциональном производимому изменению; 2) процессы, для течения которых не требуется затраты работы извне и в результате которых не может быть получена работа против внешних сил; 3) процессы, которые могут протекать самопроизвольно, т.е. без затраты работы извне, причем в результате их может быть получена работа против внешних сил в количестве, пропорциональном произведенному изменению. Примерами процессов первой группы может служить поднятие какого-либо тела на более высокий уровень, разложение воды действием электрического тока и т.д. Примерами процессов второй группы являются передвижение шара по строго горизонтальной плоскости или качание маятника без трения. К третьей группе принадлежат такие процессы, как опускание груза на более низкий уровень, взаимная нейтрализация сильной кислоты сильным основанием, любая реакция, используемая в работающем гальваническом элементе, сгорание горючего, взрыв, ржавление железа и т.д. Процессы этой группы называют положительными, в отличие от процессов первой группы, которые называют отрицательными. Основными положениями первого закона, как мы видели, являются утверждения о постоянстве количества внутренней энергии, содержащейся в изолированной системе, и об эквивалентности различных форм энергии, а также соотношения, связывающие изменения внутренней энергии системы с количеством поступившей теплоты и произведенной работы. При этом первый закон не касается характера, возможности и направления тех процессов, при которых могут или будут происходить те или иные превращения энергии. Другими словами, с точки зрения первого начала термодинамики все процессы, происходящие без нарушения закона сохранения энергии, возможны. Однако опыт показывает, что самопроизвольные процессы в природе протекают только в определенных направлениях и до определенного предела. Поэтому первое начало термодинамики необходимо дополнить началом, которое позволяло бы судить о направлениях самопроизвольных процессов и пределах их протекания. Таким началом является эмпирический закон, установленный на основании большого человеческого опыта. Справедливость этого закона подтверждается тем, что ни одно из его следствий не находится в противоречии с опытом. Второе начало термодинамики тесно связано с существованием необратимых процессов. Прежде всего познакомимся с понятиями необратимые и обратимые процессы в термодинамическом смысле. 3.2. Обратимые и необратимые процессы Для выяснения понятий обратимый и необратимый процесс в термодинамическом смысле рассмотрим изотермическое расширение 1 моля идеального газа. Допустим, что 1 моль идеального газа находится в цилиндре, снабженном невесомым поршнем, который может передвигаться вдоль стенок без трения. Стенки цилиндра обладают идеальной теплопроводностью, т.е. во время процесса температура не меняется. В начальный момент (рис. 3.1) газ занимает объем V1и находится под давлением Р1. На графике такое состояние обозначено как начальное состояние 1.
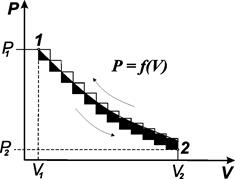
Рис. 3.1. Соотношение между Рвнеш и V, позволяющее определять работу расширения для газа, расширяющегося против постоянного внешнего давления Р2 и против переменного давления Р Начнем изменять давление бесконечно малыми шагами. Если оно будет падать, то объем будет возрастать также бесконечно малыми шагами. Таким путем можно перейти из состояния 1 в состояние 2, в котором газ будет иметь давление Р2и объем V2. Графически этот бесконечно медленный процесс изображается плавной кривой 2. Работа, совершаемая системой, ограничена изотермой с ординатами Р1и Р2и отрезком на оси абсцисс V2– V1. Это работа расширения газа. Обозначим ее W1-2. Представим себе обратный процесс, в котором мы будем путем бесконечно малого увеличения давления сжимать газ. В конечном счете мы сможем вернуть его в первоначальное состояние 1. Графически этот процесс будет описываться той же плавной кривой 2-1, но протекать в обратном направлении. В этом случае система при переходе из конечного состояния в начальное будет проходить через те же промежуточные состояния давления и объема как в прямом, так и в обратном процессах. Изменения происходили на бесконечно малые величины, и система в каждый момент времени находилась в равновесном состоянии. В этом случае работа, которую совершает система в обратном процессе W2-1,будет равной, но обратной по знаку работе прямого процесса: W12 = - W2-1; W12+ W2-1 = 0. (3.1) Следовательно, обратимый процесс – это процесс, в результате которого система может возвратиться в исходное состояние без изменений в окружающей среде. Значит, обратимые процессы протекают с бесконечно малыми скоростями. Только при этих условиях система в каждый момент времени будет находиться в состоянии, бесконечно мало отличающемся от равновесного. такие процессы называют равновесными, или квазистатическими. Проведем процесс расширения 1 моль газа с конечной скоростью. при изменении давления на конечную величину (нижняя кривая) объем газа увеличивается также на конечную величину. Последовательно перейдем из состояния 1 в состояние 2. Графически этот процесс изображен ломаной линией. Работа расширения, которую при этом совершает газ, численно равна площади под ломаной линией. Она меньше, чем в предыдущем случае. Проведем процесс в обратном направлении. Здесь также давление будет увеличиваться на конечную величину (верхняя ломаная линия). Объем газа уменьшается и через некоторое время достигает равновесного состояния. Работа, которую при этом производит внешняя среда (работа сжатия), численно равна площади, ограниченной верхней ломаной линией, двумя ординатами Р1и Р2и отрезком на оси абсцисс V1– V2. Сопоставим диаграммы сжатия и расширения и отметим, что при изменении состояния газа с конечной скоростью работа обратного процесса по абсолютной величине больше работы прямого процесса: |W1-2| <|-W2-1|, (3.2) W1-2+ W2-1< 0. (3.3) Возвращение системы из конечного состояния в начальное происходит по другому пути, и в окружающей среде остаются какие-то изменения. Необратимый процесс – это процесс, после которого система не может возвратиться в исходное состояние без изменений в окружающей среде. При протекании необратимого процесса в каждый момент времени система не находится в состоянии равновесия. Такие процессы называются неравновесными. Вывод: Все самопроизвольные процессы протекают с конечными скоростями и поэтому являются необратимыми (неравновесными) процессами. Вывод: Работа, совершаемая системой в обратимом процессе, всегда больше, чем в необратимом: Wобр> Wнеобр. (3.4) Все реальные процессы в той или иной мере могут приближаться к обратимым.Работа, производимая системой, достигает максимального значения, если система совершает обратимый процесс: Wобр= Wmax. (3.5) Работу, производимую системой при переходе из одного состояния в другое, в общем случае можно представить как сумму работы расширения и других видов работы (работы против электрических, поверхностных, гравитационных и т.п. сил).Сумму всех видов работы, производимой системой за вычитом работы расширения, называют полезной работой. Если переход системы из состояния 1 в состояние 2 был осуществлен обратимо, то работа этого процесса будет максимальной (Wmax), а работа за вычетом работы расширения – максимальной полезной работой (W'max): Wmax= W'max+ pD V; (3.6) W'max= Wmax- pD V. (3.7) 3.3. Формулировка и математическое выражение Установление второго начала термодинамики связано с исследованиями французского военного инженера С. Карно (1824 г.) принципов действия тепловых двигателей, т.е. машин, превращающих теплоту в работу, с целью повышения их коэффициента полезного действия (КПД). Окончательно второе начало термодинамики было сформулировано Р. Клаузиусом в 1850 году и У. Томпсоном (лордом Кельвином) в 1851 году. Существует несколько эквивалентных формулировок второго начала термодинамики: 1. Невозможен самопроизвольный переход тепла от тела менее нагретого к телу более нагретому (Р. Клаузис). 2. Невозможно превратить теплоту в работу, не производя никакого другого действия, кроме охлаждения источника тепла (У. Томпсон, М. Планк). 3. Вечный двигатель второго рода невозможен (В. Оствальд). Вечным двигателем второго рода называют такую машину, единственным результатом действия которой было бы получение работы за счет теплоты окружающей среды. В любой системе два произвольно выбранные состояния (1” и 2”) различаются тем, что процесс перехода из состояния 1 в состояние 2 протекает самопроизвольно, а обратный процесс перехода из состояния 2 в состояние 1 самопроизвольно не идет. Отсюда можно заключить, что существует какой-то объективный критерий, позволяющий установить принципиальное различие между этими двумя состояниями системы. Рассмотрим изолированную систему, состоящую из теплового резервуара, 1 моля идеального газа, заключенного в цилиндре с подвижным поршнем, и устройства, позволяющего за счет перемещения поршня совершать работу (рис. 3.2).
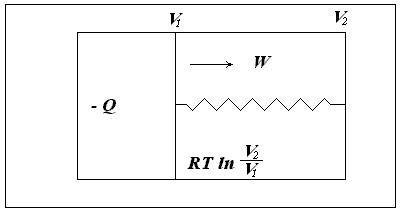
Рис. 3.2. Изолированная система, состоящая из теплового резервуара, 1 моля идеального газа, заключенного в цилиндре с подвижным поршнем, и устройства, позволяющего за счет перемещения поршня совершать работу Предположим, что газ обратимо расширяется от объема V1до V2и совершает работу W1. Энергия на совершение работы передается в форме тепла из резервуара. совершаемая газом работа равна полученной от резервуара энергии Q1:
Функция
Из равенства (3.9) видно, что изменения, происходящие в изолированной системе при протекании обратимого процесса, могут быть охарактеризованы величиной
При необратимом (самопроизвольном) расширении идеального газа от V1до V2(например, в вакууме, рис. 3.3) процесс происходит без совершения газом работы, так как Р=О и, соответственно, передача энергии от резервуара не происходит: Q=0, т.е. изменение внутренней энергии (DU) для газа равно нулю.
Рис. 3.3. При самопроизвольном расширении идеального газа от V1 до V2в вакууме процесс происходит без совершения газом работы, так как Р = О и, Однако состояние газа в резервуаре изменилось на величину
Таким образом, протекание самопроизвольного процесса в изолированной системе в общем случае связано с возрастанием некоего параметра состояния системы. Этот параметр получил название энтропии. Из примера следует, что самопроизвольно в изолированной системе протекают те процессы, которые приводят к возрастанию энтропии системы. Если энтропия системы в исходном состоянии может быть выражена как S1 = R ln V1, а в конечном состоянии S2 = R ln V2, то изменение энтропии в результате протекания обратимого процесса следующее: DS = S2- S1= R ln V2/V1, или DS(обр.процесс) = Соответственно для необратимого процесса (самопроизвольного) DS(необр.процесс) > Справедливость последнего выражения следует из первого начала термодинамики. В соответствии с I началом: DU = Q – W. (3.14) Переведем систему из состояния 1 в состояние 2 обратимым и необратимым путем: DU(обр)=Qобр – Wобр, (3.15) DU(необр) = Qнеобр – Wнеобр. (3.16) Если DU является функцией состояния, то DU(обр) = DU(необр). Известно, что Wобр > Wнеобр, следовательно Qобр > Qнеобр. DS не зависит от пути процесса, так как является функцией состояния, т.е. DS(обр)= DS(необр).Тогда
или в общем случае
Знак равенства относится к обратимым, неравенства – к необратимым процессам. 3.4. Изменение энтропии изолированной системы Для изолированной системы Q = 0, так как система не обменивается с окружающей средой ни веществом, ни энергией, и DS ³ 0. (3.20) Для обратимого процесса DS = 0; для необратимого энтропия системы увеличивается: DS > 0. (3.21) Какие бы процессы ни протекали в изолированной системе, ее энтропия не может уменьшаться. Так как самопроизвольные процессы в изолированных системах идут с увеличением энтропии, то при достижении равновесия энтропия изолированной системы будет максимальной, а ее изменение равно нулю: Sравн.= Smax, (3.22) DS = 0. (3.23) Уравнения (3.22; 3.23) – критерии равновесия изолированных систем. 3.5. Статистическая природа второго начала В то время как первое начало термодинамики является всеобщим законом природы, не знающим ограничений и применимым к любым системам, второе начало термодинамики представляет собой статистический закон, справедливый для макроскопических систем, состоящих из очень большого числа частиц (молекул, атомов, ионов), для которых применимы физические понятия, имеющие статистическую природу (например, как температура, давление). Известно, что состояние и свойства любой макроскопической системы, состоящей из совокупности большого числа частиц, могут быть описаны с помощью статистической механики. Сущность статистического описания макросистем состоит в применении к совокупности большого числа частиц основных положений теории вероятности, а к отдельным частицам – законов классической механики. С точки зрения статистической механики второе начало термодинамики, как это впервые было показано Л. Больцманом, сводится к утверждению: все самопроизвольные процессы в макроскопических системах протекают в направлении от менее вероятного к более вероятному состоянию системы. Таким образом, процессы, запрещенные вторым началом, например самопроизвольный переход тепла от менее нагретого тела к более нагретому, оказываются не невозможными, а крайне маловероятными, вследствие чего они не наблюдаются. Любое состояние системы характеризуется определенной термодинамической вероятностью, и чем больше последняя, тем ближе система к состоянию равновесия. В состоянии равновесия система обладает максимальной термодинамической вероятностью. Л. Больцман предложил следующее уравнение, устанавливающее связь между энтропией S и термодинамической вероятностью w: S = k ln w, (3.24) где k – постоянная Больцмана, численно равная отношению газовой постоянной R к числу Авогадрo NA, т.е. Статистическая термодинамика показывает, что энтропия может рассматриваться как сумма составляющих, относящихся к различным формам движения частиц. Принято группировать их по характеру движения частиц, рассматривая следующие составляющие энтропии: энтропию поступательного движения молекул (Sпост.), энтропию вращательного движения молекул (Sвращ.), энтропию вращательного движения атомов и атомных групп, содержащихся в молекуле (Sвн.вращ.), энтропию колебательного движения атомов и атомных групп (Sкол) и энтропию движения электронов (Sэл.). Таким образом, энтропию можно представить как сумму следующих составляющих: S = Sпост +Sвращ + Sвн.вращ + Sкол + Sэл. (3.25) При этом некоторые из них можно рассматривать как сумму более частных составляющих. Так, Sкол. является суммой составляющих, относящихся к различным видам колебаний. При рассмотрении обычных химических процессов не учитывают составляющие энтропии, связанные с состоянием атомных ядер (спиновой эффект) и с изотопным эффектом. Для каждого данного вещества энтропия возрастает при всех процессах, вызываемых движением частиц (испарение, плавление, расширение газов, диффузия и пр.). энтропия возрастает при ослаблении связей между атомами в молекулах и при разрыве их, т.е. диссоциации молекул на атомы или атомные группы. Наоборот, с упрочением связей уменьшается энтропия. Вместе с тем второе начало термодинамики не настолько простое в смысле его применения. Рассмотрим, например, условия и задачу, которая была решена Клаузиусом в середине XIX века следующим образом. Если вселенная является изолированной системой, и энтропия в обратимых процессах не меняется, а в необратимых только возрастает, то это возрастание должно приводить к постепенному выравниванию температуры во всех ее частях. В плане вселенной это должно привести в конце концов к полному выравниванию температуры, т.е. к «тепловой смерти». Правомерность такого вывода рассматривается с разных сторон такими учеными, как М. Смолуховский, Я. Ван-дер-Ваальс и др. Можем ли мы в настоящее время на основе тех знаний, которые у нас есть, его оспорить? Лекция 4. 4.1. Формулировка третьего начала термодинамики В 1912 году М. Планк высказал постулат: при абсолютном нуле энтропия правильно образованного кристалла чистого вещества равна нулю. Справедливость постулата Планка, называемого третьим началом термодинамики, следует из экспериментальных данных о зависимости теплоемкости кристаллических веществ от температуры, а также из статистического характера второго начала термодинамики. При абсолютном нуле данное макросостояние кристалла чистого вещества, кристаллическая решетка которого не имеет каких-либо дефектов, предельно упорядочено и может быть реализовано единственным способом. Следовательно, термодинамическая вероятность при абсолютном нуле равна 1: S = k ln 1 S = 0. (4.1) На основании постулата Планка можно вычислить абсолютное значение энтропии. Если Интегрируя последнее уравнение в пределах от абсолютного нуля до Т, получим:
или ST = 4.2. Абсолютные и стандартные значения энтропии Энтропию ST называют абсолютной энтропией. Она численно равна изменению энтропии при равновесном переходе 1 моля кристаллического вещества от абсолютного нуля до данной температуры. Вычисление абсолютной энтропии по уравнению (4.3) возможно лишь в том случае, если известна зависимость теплоемкости данного вещества от температуры. Абсолютную энтропию тела в стандартном состоянии при данном Т называют стандартной энтропией и обозначают через S0T; чаще всего Т=298.15 °К и ее обозначают через S0298: S0298 = Зная стандартную энтропию, можно вычислить значение абсолютной энтропии данного вещества при любой температуре Т: ST = S0298 + Таблицы стандартных значений энтропий играют существенную роль при расчетах констант химических реакционных равновесий и определении направления протекания химических реакций и других процессов. Необходимо подчеркнуть, что третье начало термодинамики дает возможность вычислить абсолютное значение энтропии различного рода веществ при данном состоянии, тогда как для других термодинамических функций (U, H) могут быть определены только их изменения при переходе данной системы из одного состояния в другое. 4.3. Расчеты изменения энтропии в различных процессах Методов непосредственного измерения энтропии не существует, но можно рассчитать изменение этой функции при различного рода процессах, пользуясь математическим выражением второго начала термодинамики для обратимых процессов. Так как энтропия является функцией состояния, то ее изменение при переходе системы из начального состояния в конечное будет одним и тем же независимо от того, совершается ли этот переход обратимым или необратимым путем. 
Не нашли, что искали? Воспользуйтесь поиском по сайту: ©2015 - 2024 stydopedia.ru Все материалы защищены законодательством РФ.
|
 . (3.8)
. (3.8) определяется не только изменением объема, но и температурой. Разделим обе части уравнения на Т:
определяется не только изменением объема, но и температурой. Разделим обе части уравнения на Т: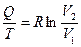 . (3.9)
. (3.9) , которая определяется только исходным (V1) и конечным (V2) состоянием системы. Увеличение параметра
, которая определяется только исходным (V1) и конечным (V2) состоянием системы. Увеличение параметра 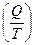 , т.е.
, т.е.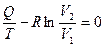 . (3.10)
. (3.10)
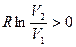 , так как
, так как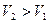 . (3.11)
. (3.11) . (3.12)
. (3.12)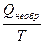 . (3.13)
. (3.13)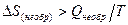 (3.17)
(3.17)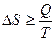 ; (3.18)
; (3.18)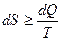 . (3.19)
. (3.19) ; w – термодинамическая вероятность системы, т.е. число микросостояний, которыми можно осуществить данное макросостояние системы.
; w – термодинамическая вероятность системы, т.е. число микросостояний, которыми можно осуществить данное макросостояние системы.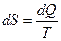 , а
, а 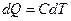 , то
, то 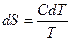 , где С – молярная теплоемкость данного вещества.
, где С – молярная теплоемкость данного вещества.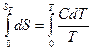 (4.2)
(4.2)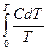 . (4.3)
. (4.3) . (4.4)
. (4.4)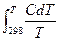 . (4.5)
. (4.5)